V-ATPase
ATPase. For the gastric H+
/K+
ATPase, see Hydrogen potassium ATPase. For the plant/fungal plasma membrane H+
ATPase, see Proton ATPase.
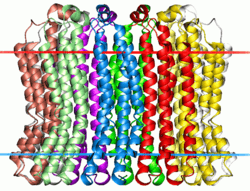
|
Membrane-spanning region of the V-type sodium ATPase from Enterococcus hirae. Calculated hydrocarbon boundaries of the lipid bilayer are shown by red and blue dots | |||||||||
| Identifiers | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Symbol | ATP-synt_C | ||||||||
| Pfam | PF00137 | ||||||||
| InterPro | IPR002379 | ||||||||
| PROSITE | PDOC00526 | ||||||||
| SCOP | 1aty | ||||||||
| SUPERFAMILY | 1aty | ||||||||
| OPM superfamily | 5 | ||||||||
| OPM protein | 2bl2 | ||||||||
| |||||||||
| V-ATPase_C | |||||||||
|---|---|---|---|---|---|---|---|---|---|
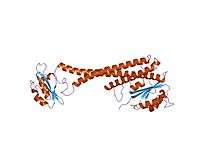 crystal structure of subunit C (vma5p) of the yeast v-atpase | |||||||||
| Identifiers | |||||||||
| Symbol | V-ATPase_C | ||||||||
| Pfam | PF03223 | ||||||||
| InterPro | IPR004907 | ||||||||
| SCOP | 1u7l | ||||||||
| SUPERFAMILY | 1u7l | ||||||||
| |||||||||
| V_ATPase_I | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| Symbol | V_ATPase_I | ||||||||
| Pfam | PF01496 | ||||||||
| InterPro | IPR002490 | ||||||||
| SCOP | 3rrk | ||||||||
| SUPERFAMILY | 3rrk | ||||||||
| TCDB | 3.A.2 | ||||||||
| |||||||||
| vATP-synt_E | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| Symbol | vATP-synt_E | ||||||||
| Pfam | PF01991 | ||||||||
| Pfam clan | CL0255 | ||||||||
| InterPro | IPR002842 | ||||||||
| |||||||||
| vATP-synt_AC39 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
 crystal structure of subunit C (yeast subunit d) of v-atpase | |||||||||
| Identifiers | |||||||||
| Symbol | vATP-synt_AC39 | ||||||||
| Pfam | PF01992 | ||||||||
| InterPro | IPR002843 | ||||||||
| SCOP | 1r5z | ||||||||
| SUPERFAMILY | 1r5z | ||||||||
| |||||||||
| V-ATPase_H_N | |||||||||
|---|---|---|---|---|---|---|---|---|---|
 crystal structure of the regulatory subunit H of the v-type atpase of saccharomyces cerevisiae | |||||||||
| Identifiers | |||||||||
| Symbol | V-ATPase_H_N | ||||||||
| Pfam | PF03224 | ||||||||
| Pfam clan | CL0020 | ||||||||
| InterPro | IPR004908 | ||||||||
| SCOP | 1ho8 | ||||||||
| SUPERFAMILY | 1ho8 | ||||||||
| |||||||||
| V-ATPase_G | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| Symbol | V-ATPase_G | ||||||||
| Pfam | PF03179 | ||||||||
| Pfam clan | CL0255 | ||||||||
| InterPro | IPR005124 | ||||||||
| |||||||||
Vacuolar-type H+
-ATPase (V-ATPase) is a highly conserved evolutionarily ancient enzyme with remarkably diverse functions in eukaryotic organisms.[1] V-ATPases acidify a wide array of intracellular organelles and pump protons across the plasma membranes of numerous cell types. V-ATPases couple the energy of ATP hydrolysis to proton transport across intracellular and plasma membranes of eukaryotic cells. It is generally seen as the polar opposite of ATP Synthase because ATP Synthase is a proton channel that uses the energy from a proton gradient to produce ATP. V-ATPase however, is a proton pump that uses the energy from ATP hydrolysis to produce a proton gradient.
Roles played by V-ATPases
V-ATPases are found within the membranes of many organelles, such as endosomes, lysosomes, and secretory vesicles, where they play a variety of roles crucial for the function of these organelles. For example, the proton gradient across the yeast vacuolar membrane generated by V-ATPases drives calcium uptake into the vacuole through an H+
/Ca2+
antiporter system (Ohya, 1991). In synaptic transmission in neuronal cells, V-ATPase acidifies synaptic vesicles.[2] Norepinephrine enters vesicles by V-ATPase.
V-ATPases are also found in the plasma membranes of a wide variety of cells such as intercalated cells of the kidney, osteoclasts (bone resorbing cells), macrophages, neutrophils, sperm, midgut cells of insects, and certain tumor cells.[3] Plasma membrane V-ATPases are involved in processes such as pH homeostasis, coupled transport, and tumor metastasis. V-ATPases in the acrosomal membrane of sperm acidify the acrosome. This acidification activates proteases required to drill through the plasma membrane of the egg. V-ATPases in the osteoclast plasma membrane pump protons onto the bone surface, which is necessary for bone resorption. In the intercalated cells of the kidney, V-ATPases pump protons into the urine, allowing for bicarbonate reabsorption into the blood.
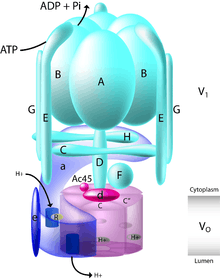
V-ATPase structure
The yeast V-ATPase is the best characterized. There are at least 13 subunits identified to form a functional V-ATPase complex, which consists of two domains. The subunits belong to either the Vo domain (membrane associated subunits, lowercase letters on the figure) or the V1 domain (peripherally associated subunits, uppercase letters on the figure).
The V1 includes 8 subunits, A-H, with three copies of the catalytic A and B subunits, three copies of the stator subunits E and G, and one copy of the regulatory C and H subunits. In addition, the V1 domain also contains the subunits D and F, which form a central rotor axle (Kitagawa et al., 2008). The V1 domain contains tissue-specific subunit isoforms including B, C, E, and G. Mutations to the B1 isoform result in the human disease distal renal tubular acidosis and sensorineural deafness.
The Vo domain contains 6 different subunits, a, d, c, c', c", and e, with the stoichiometry of the c ring still a matter of debate with a decamer being postulated for the tobacco Hornworm Manduca sexta V-ATPase. The mammalian Vo domain contains tissue-specific isoforms for subunits a and d, while yeast V-ATPase contains two organelle-specific subunit isoforms of a, Vph1p, and Stv1p. Mutations to the a3 isoform result in the human disease infantile malignant osteopetrosis, and mutations to the a4 isoform result in distal renal tubular acidosis, in some cases with sensorineural deafness.
The V1 domain is responsible for ATP hydrolysis, whereas the Vo domain is responsible for proton translocation. ATP hydrolysis at the catalytic nucleotide binding sites on subunit A drives rotation of a central stalk composed of subunits D and F, which in turn drives rotation of a barrel of c subunits relative to the a subunit. The complex structure of the V-ATPase has been revealed through the structure of the M. Sexta and Yeast complexes that were solved by single-particle cryo-EM and negative staining, respectively (Muench 2009, Diepholz 2008, Zhang 2008). These structures have revealed that the V-ATPase has a 3-stator network, linked by a collar of density formed by the C, H, and a subunits, which, while dividing the V1 and V0 domains, make no interactions with the central rotor axle formed by the F, D, and d subunits. Rotation of this central rotor axle caused by the hydrolysis of ATP within the catalytic AB domains results in the movement of the barrel of c subunits past the a subunit, which drives proton transport across the membrane. A stoichiometry of two protons translocated for each ATP hydrolyzed has been proposed by (Johnson, 1982).
In addition to the structural subunits of yeast V-ATPase, associated proteins that are necessary for assembly have been identified. These associated proteins are essential for Vo domain assembly and are termed Vma12p, Vma21p, and Vma22p (Hirata, 1993; Ho, 1993; Hill, 1994; Jackson, 1997). Two of the three proteins, Vma12p and Vma22p, form a complex that binds transiently to Vph1p (subunit a) to aid its assembly and maturation (Hill, 1994; Hill, 1995; Graham, 1998; Graham, 2003). Vma21p coordinates assembly of the Vo subunits as well as escorting the Vo domain into vesicles for transport to the Golgi (Malkus, 2004).
V1
The V1 domain of the V-ATPase is the site of ATP hydrolysis. This soluble domain consists of a hexamer of alternating A and B subunits, a central rotor D, peripheral stators G and E, and regulatory subunits C and H. Hydrolysis of ATP drives a conformational change in the six A|B interfaces and with it rotation of the central rotor D. Unlike with the ATP synthase, the V1 domain is not an active ATPase when dissociated.
Subunit C
In molecular biology, V-ATPase (Vacuolar-ATPase) C represents the C terminal subunit that is part of the V1 complex, and is localised to the interface between the V1 and V0 complexes.[4]
Subunit C function
The C subunit plays an essential role in controlling the assembly of V-ATPase, acting as a flexible stator that holds together the catalytic (V1) and membrane (V0) sectors of the enzyme .[5] The release of subunit C from the ATPase complex results in the dissociation of the V1 and V0 subcomplexes, which is an important mechanism in controlling V-ATPase activity in cells. Essentially, by creating a high electrochemical gradient and low pH, this powers the enzyme to create more ATP.
Subunit G
This subunit, is part of V1, and is important in V-ATPase assembly and activity.
Subunit H
This subunit is only involved in activity and not in assembly.
Vo
The Vo domain is responsible for proton translocation. Opposite the F-type [ATP_synthase], the Vo domain is transporting protons against their own concentration gradient. Rotation of the Vo domain transports the protons in movement coordinated with the V1 domain, which is responsible for ATP hydrolysis. Several subunits are present in the Vo domain to make this a functional proton translocase; they are described below.
Subunit I
In molecular biology, 116kDa subunit (or subunit a) and subunit I are found in the V0 or A0 complex of V- or A-ATPases, respectively. The 116kDa subunit is a transmembrane glycoprotein required for the assembly and proton transport activity of the ATPase complex. Several isoforms of the 116kDa subunit exist, providing a potential role in the differential targeting and regulation of the V-ATPase for specific organelles.
Subunit I function
The function of the 116-kDa subunit is not defined, but its predicted structure consists of 6–8 transmembranous sectors, suggesting that it may function similar to subunit a of FO.
Subunit d
This particular subunit is a non-integral membrane component of the membrane pore domain and is required for proper assembly of the V0 sector. It is thought to be involved in the regulated assembly of V1 subunits onto the membrane sector or alternatively may prevent the passage of protons through V0 pores.
Subunit d2
This subunit is part of the integral membrane V0 complex of vacuolar ATPase, which is responsible for acidifying intracellular compartments in eukaryotic cells. Therefore, they help provide most of the energy required for transport processes in the vacuolar system. They are thought to play a role in coupling of proton transport and ATP hydrolysis and aid the regulation of osteoclast fusion and bone formation.
Subunit c
Similar to the F-type ATP synthase, the transmembrane region of the V-ATPase includes a ring of membrane-spanning subunits that are primarily responsible for proton translocation. Dissimilar from the F-type ATP synthase, however, the V-ATPase has multiple related subunits in the c-ring; in fungi such as yeast there are three related subunits (of varied stoichiometry) and in most other eukaryotes there are two.
V-ATPase assembly
Yeast V-ATPases fail to assemble when any of the genes that encode subunits are deleted except for subunits H and c" (Whyteside, 2005; Forgac, 1999; Stevens, 1997). Without subunit H, the assembled V-ATPase is not active (Ho, 1993; Parra, 2000) and the loss of the c" subunit results in uncoupling of enzymatic activity (Whyteside, 2005).
The precise mechanisms by which V-ATPases assembly are still controversial, with evidence suggesting two different possibilities. Mutational analysis and in vitro assays have shown that preassembled Vo and V1 domains can combine to form one complex in a process called independent assembly. Support for independent assembly includes the findings that the assembled Vo domain can be found at the vacuole in the absence of the V1 domain, whereas free V1 domains can be found in the cytoplasm and not at the vacuole (Kane, 1995; Sumner, 1995). In contrast, in vivo pulse-chase experiments have revealed early interactions between Vo and V1 subunits, to be specific, the a and B subunits, suggesting that subunits are added in a step-wise fashion to form a single complex in a concerted assembly process (Kane, 1999).
V-ATPase evolution
A relatively new technique called ancestral gene resurrection has shed new light on the evolutionary history of the V-ATPase. It has been shown how the V-ATPase structure of the ancestral form consisting of two different proteins evolves into the fungi version with three different proteins.[6][7][8]
Regulation of V-ATPase activity
In vivo regulation of V-ATPase activity is accomplished by reversible dissociation of the V1 domain from the Vo domain. After initial assembly, both the insect Manduca sexta and yeast V-ATPases can reversibly disassemble into free Vo and V1 domains after a 2- to 5-minute deprivation of glucose (Kane, 1995). Reversible disassembly may be a general mechanism of regulating V-ATPase activity, since it exists in yeast and insects. Reassembly is proposed to be aided by a complex termed RAVE (regulator of H+
-ATPase of vacuolar and endosomal membranes) (Kane and Smardon, 2003). Dissasembly and reassembly of V-ATPases does not require new protein synthesis but does need an intact microtubular network (Holliday, 2000).
Human diseases
Osteopetrosis
Osteopetrosis is generic name that represents a group of heritable conditions in which there is a defect in osteoclastic bone resorption. Both dominant and recessive osteopetrosis occur in humans {Michigami, 2002; Frattini, 2000}. Autosomal dominant osteopetrosis shows mild symptoms in adults experiencing frequent bone fractures due to brittle bones {Michigami, 2002}. A more severe form of osteopetrosis is termed autosomal recessive infantile malignant osteopetrosis {Frattini, 2000; Sobacchi, 2001; Fasth, 1999}. Three genes that are responsible for recessive osteopetrosis in humans have been identified. They are all directly involved in the proton generation and secretion pathways that are essential for bone resorption. One gene is carbonic anhydrase II (CAII), which, when mutated, causes osteopetrosis with renal tubular acidosis(type 3) {Sly, 1983}. Mutations to the chloride channel ClC7 gene also lead to both dominant and recessive osteopetrosis {Michigami, 2002}. Approximately 50% of patients with recessive infantile malignant osteopetrosis have mutations to the a3 subunit isoform of V-ATPase {Sobacchi, 2001; Kornak, 2000; Frattini, 2003}. In humans, 26 mutations have been identified in V-ATPase subunit isoform a3, found in osteoclasts, that result in the bone disease autosomal recessive osteopetrosis {Frattini, 2000; Kornak, 2000; Sobacchi, 2001; Susani, 2004}.
Distal renal tubular acidosis (dRTA)
The importance of V-ATPase activity in renal proton secretion is highlighted by the inherited disease distal renal tubular acidosis. In all cases, renal tubular acidosis results from a failure of the normal renal mechanisms that regulate systemic pH. There are four types of renal tubular acidosis. Type 1 is distal renal tubular acidosis and results from a failure of the cortical collecting duct to acidify the urine below pH 5. {Alper, 2002}. Some patients with autosomal recessive dRTA also have sensorineural hearing loss {Karet, 1999}. Inheritance of this type of RTA results from either mutations to V-ATPase subunit isoform B1 or isoform a4 or mutations of band 3 (also called AE1), a Cl-/HCO3- exchanger {Stehberger, 2003; Karet, 1999; Karet, 1998}. Twelve different mutations to V-ATPase isoform B1 (Stover, 2002) and twenty-four different mutations in a4 lead to dRTA {Smith, 2000; Karet, 1999; Stover, 2005}. Reverse transcription polymerase chain reaction studies have shown expression of the a4 subunit in the intercalated cell of the kidney and in the cochlea {Stover, 2002}. dRTA caused by mutations in the a4 subunit gene in some cases can be associated with deafness due to a failure to normally acidify the endolymph of the inner ear {Stehberger, 2003}.
X-linked myopathy with excessive autophagy (XMEA)
X-linked myopathy with excessive autophagy is a rare genetic disease resulting from mutations in the VMA21 gene.[9] The disease has a childhood onset and results in a slowly progressive muscle weakness, typically beginning in the legs, and some patients can eventually require wheelchair assistance with advanced age. The Vma21 protein assists in assembly of the V-ATPase, and XMEA associated mutations result in decreased activity of the V-ATPase and increased lysosomal pH.[9]
Nomenclature
The term Vo has a lowercase letter "o" (not the number "zero") in subscript. The "o" stands for oligomycin. It is worth noting that the human gene notations at NCBI designate it as "zero" rather than the letter "o". For example, the gene for the human c subunit of Vo is listed in NCBI gene database as "ATP6V0C" (with a zero), rather than "ATP6VOC" (with an "o").
See also
References
- ↑ Nelson N, Perzov N, Cohen A, Hagai K, Padler V, Nelson H; Perzov; Cohen; Hagai; Padler; Nelson (1 January 2000). "The cellular biology of proton-motive force generation by V-ATPases". J. Exp. Biol. 203 (Pt 1): 89–95. PMID 10600677.
- ↑ Wienisch M, Klingauf J; Klingauf (August 2006). "Vesicular proteins exocytosed and subsequently retrieved by compensatory endocytosis are nonidentical". Nat. Neurosci. 9 (8): 1019–27. doi:10.1038/nn1739. PMID 16845386.
- ↑ Izumi H; Torigoe T; Ishiguchi H; et al. (December 2003). "Cellular pH regulators: potentially promising molecular targets for cancer chemotherapy". Cancer Treat. Rev. 29 (6): 541–9. doi:10.1016/S0305-7372(03)00106-3. PMID 14585264.
- ↑ Inoue T, Forgac M; Forgac (July 2005). "Cysteine-mediated cross-linking indicates that subunit C of the V-ATPase is in close proximity to subunits E and G of the V1 domain and subunit a of the V0 domain". J. Biol. Chem. 280 (30): 27896–903. doi:10.1074/jbc.M504890200. PMID 15951435.
- ↑ Drory O, Frolow F, Nelson N; Frolow; Nelson (December 2004). "Crystal structure of yeast V-ATPase subunit C reveals its stator function". EMBO Rep. 5 (12): 1148–52. doi:10.1038/sj.embor.7400294. PMC 1299189
 . PMID 15540116.
. PMID 15540116. - ↑ Resurrecting extinct proteins shows how a machine evolves. (Accessed 2012-01-11).
- ↑ Finnigan and Hanson-Smith, et al. Evolution of increased complexity in a molecular machine. Nature (2012). doi:10.1038/nature10724. (Accessed 2012-01-11)
- ↑ Snapshot view of the V-ATPase molecular machine: animals vs. fungi, University of Oregon (Accessed 2012-01-11)
- 1 2 Ramachandran, N; Munteanu, I; Wang, P; Ruggieri, A; Rilstone, J. J.; Israelian, N; Naranian, T; Paroutis, P; Guo, R; Ren, Z. P.; Nishino, I; Chabrol, B; Pellissier, J. F.; Minetti, C; Udd, B; Fardeau, M; Tailor, C. S.; Mahuran, D. J.; Kissel, J. T.; Kalimo, H; Levy, N; Manolson, M. F.; Ackerley, C. A.; Minassian, B. A. (2013). "VMA21 deficiency prevents vacuolar ATPase assembly and causes autophagic vacuolar myopathy". Acta Neuropathologica. 125 (3): 439–57. doi:10.1007/s00401-012-1073-6. PMID 23315026.
- Alper SL (2002). "Genetic diseases of acid-base transporters". Annu. Rev. Physiol. 64: 899–923. doi:10.1146/annurev.physiol.64.092801.141759. PMID 11826292.
- Diepholz M, Börsch M, Böttcher B; Börsch; Böttcher (October 2008). "Structural organization of the V-ATPase and its implications for regulatory assembly and disassembly". Biochem. Soc. Trans. 36 (Pt 5): 1027–31. doi:10.1042/BST0361027. PMID 18793183.
- Fasth A, Porras O; Porras (1999). "Human malignant osteopetrosis: pathophysiology, management and the role of bone marrow transplantation". Pediatr Transplant. 3 (Suppl 1): 102–7. doi:10.1034/j.1399-3046.1999.00063.x. PMID 10587979.
- Forgac M (January 1999). "The vacuolar H+
-ATPase of clathrin-coated vesicles is reversibly inhibited by S-nitrosoglutathione". J. Biol. Chem. 274 (3): 1301–5. doi:10.1074/jbc.274.3.1301. PMID 9880499. - Frattini A; Orchard PJ; Sobacchi C; et al. (July 2000). "Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis". Nat. Genet. 25 (3): 343–6. doi:10.1038/77131. PMID 10888887.
- Frattini A; Pangrazio A; Susani L; et al. (October 2003). "Chloride channel ClCN7 mutations are responsible for severe recessive, dominant, and intermediate osteopetrosis". J. Bone Miner. Res. 18 (10): 1740–7. doi:10.1359/jbmr.2003.18.10.1740. PMID 14584882.
- Graham LA, Hill KJ, Stevens TH; Hill; Stevens (July 1998). "Assembly of the yeast vacuolar H+
-ATPase occurs in the endoplasmic reticulum and requires a Vma12p/Vma22p assembly complex". J. Cell Biol. 142 (1): 39–49. doi:10.1083/jcb.142.1.39. PMC 2133036. PMID 9660861. - Graham LA, Flannery AR, Stevens TH; Flannery; Stevens (August 2003). "Structure and assembly of the yeast V-ATPase". J. Bioenerg. Biomembr. 35 (4): 301–12. doi:10.1023/A:1025772730586. PMID 14635776.
- Kitagawa N, Mazon H, Heck AJ, Wilkens S; Mazon; Heck; Wilkens (February 2008). "Stoichiometry of the peripheral stalk subunits E and G of yeast V1-ATPase determined by mass spectrometry". J. Biol. Chem. 283 (6): 3329–37. doi:10.1074/jbc.M707924200. PMID 18055462.
- Hill KJ, Stevens TH; Stevens (September 1994). "Vma21p is a yeast membrane protein retained in the endoplasmic reticulum by a di-lysine motif and is required for the assembly of the vacuolar H+
-ATPase complex". Mol. Biol. Cell. 5 (9): 1039–50. doi:10.1091/mbc.5.9.1039. PMC 301125. PMID 7841520. - Hill KJ, Stevens TH; Stevens (September 1995). "Vma22p is a novel endoplasmic reticulum-associated protein required for assembly of the yeast vacuolar H+
-ATPase complex". J. Biol. Chem. 270 (38): 22329–36. doi:10.1074/jbc.270.38.22329. PMID 7673216. - Hirata R, Umemoto N, Ho MN, Ohya Y, Stevens TH, Anraku Y; Umemoto; Ho; Ohya; Stevens; Anraku (January 1993). "VMA12 is essential for assembly of the vacuolar H+
-ATPase subunits onto the vacuolar membrane in Saccharomyces cerevisiae". J. Biol. Chem. 268 (2): 961–7. PMID 8419376. - Ho MN; Hirata R; Umemoto N; et al. (August 1993). "VMA13 encodes a 54-kDa vacuolar H+
-ATPase subunit required for activity but not assembly of the enzyme complex in Saccharomyces cerevisiae". J. Biol. Chem. 268 (24): 18286–92. PMID 8349704. - Holliday LS; Lu M; Lee BS; et al. (October 2000). "The amino-terminal domain of the B subunit of vacuolar H+
-ATPase contains a filamentous actin binding site". J. Biol. Chem. 275 (41): 32331–7. doi:10.1074/jbc.M004795200. PMID 10915794. - Jackson DD, Stevens TH; Stevens (October 1997). "VMA12 encodes a yeast endoplasmic reticulum protein required for vacuolar H+
-ATPase assembly". J. Biol. Chem. 272 (41): 25928–34. doi:10.1074/jbc.272.41.25928. PMID 9325326. - Johnson RG, Beers MF, Scarpa A; Beers; Scarpa (September 1982). "H+
ATPase of chromaffin granules. Kinetics, regulation, and stoichiometry". J. Biol. Chem. 257 (18): 10701–7. PMID 6213624. - Kane PM (July 1995). "Disassembly and reassembly of the yeast vacuolar H+
-ATPase in vivo". J. Biol. Chem. 270 (28): 17025–32. doi:10.1074/jbc.270.28.17025 (inactive 2015-02-01). PMID 7622524. - Kane PM, Tarsio M, Liu J; Tarsio; Liu (June 1999). "Early steps in assembly of the yeast vacuolar H+
-ATPase". J. Biol. Chem. 274 (24): 17275–83. doi:10.1074/jbc.274.24.17275. PMID 10358087. - Kane PM, Smardon AM; Smardon (August 2003). "Assembly and regulation of the yeast vacuolar H+
-ATPase". J. Bioenerg. Biomembr. 35 (4): 313–21. doi:10.1023/A:1025724814656. PMID 14635777. - Karet FE; Gainza; Gyory; et al. (May 1998). "Mutations in the chloride-bicarbonate exchanger gene AE1 cause autosomal dominant but not autosomal recessive distal renal tubular acidosis". Proc. Natl. Acad. Sci. U.S.A. 95 (11): 6337–42. Bibcode:1998PNAS...95.6337K. doi:10.1073/pnas.95.11.6337. PMC 27686. PMID 9600966.
- Karet FE; Finberg KE; Nelson RD; et al. (January 1999). "Mutations in the gene encoding B1 subunit of H+
-ATPase cause renal tubular acidosis with sensorineural deafness". Nat. Genet. 21 (1): 84–90. doi:10.1038/5022. PMID 9916796. - Kornak U; Schulz A; Friedrich W; et al. (August 2000). "Mutations in the a3 subunit of the vacuolar H+
-ATPase cause infantile malignant osteopetrosis". Hum. Mol. Genet. 9 (13): 2059–63. doi:10.1093/hmg/9.13.2059. PMID 10942435. - Malkus P, Graham LA, Stevens TH, Schekman R; Graham; Stevens; Schekman (November 2004). "Role of Vma21p in assembly and transport of the yeast vacuolar ATPase". Mol. Biol. Cell. 15 (11): 5075–91. doi:10.1091/mbc.E04-06-0514. PMC 524777. PMID 15356264.
- Michigami T; Kageyama T; Satomura K; et al. (February 2002). "Novel mutations in the a3 subunit of vacuolar H+
-adenosine triphosphatase in a Japanese patient with infantile malignant osteopetrosis". Bone. 30 (2): 436–9. doi:10.1016/S8756-3282(01)00684-6. PMID 11856654. - Muench SP; Huss M; Song CF; et al. (March 2009). "Cryo-electron microscopy of the vacuolar ATPase motor reveals its mechanical and regulatory complexity". J. Mol. Biol. 386 (4): 989–99. doi:10.1016/j.jmb.2009.01.014. PMID 19244615.
- Ohya Y, Umemoto N, Tanida I, Ohta A, Iida H, Anraku Y; Umemoto; Tanida; Ohta; Iida; Anraku (July 1991). "Calcium-sensitive cls mutants of Saccharomyces cerevisiae showing a Pet- phenotype are ascribable to defects of vacuolar membrane H+
-ATPase activity". J. Biol. Chem. 266 (21): 13971–7. PMID 1830311. - Parra KJ, Keenan KL, Kane PM; Keenan; Kane (July 2000). "The H subunit (Vma13p) of the yeast V-ATPase inhibits the ATPase activity of cytosolic V1 complexes". J. Biol. Chem. 275 (28): 21761–7. doi:10.1074/jbc.M002305200. PMID 10781598.
- Sly WS, Hewett-Emmett D, Whyte MP, Yu YS, Tashian RE; Hewett-Emmett; Whyte; Yu; Tashian (May 1983). "Carbonic anhydrase II deficiency identified as the primary defect in the autosomal recessive syndrome of osteopetrosis with renal tubular acidosis and cerebral calcification". Proc. Natl. Acad. Sci. U.S.A. 80 (9): 2752–6. Bibcode:1983PNAS...80.2752S. doi:10.1073/pnas.80.9.2752. PMC 393906. PMID 6405388.
- Sobacchi C; Frattini A; Orchard P; et al. (August 2001). "The mutational spectrum of human malignant autosomal recessive osteopetrosis". Hum. Mol. Genet. 10 (17): 1767–73. doi:10.1093/hmg/10.17.1767. PMID 11532986.
- Stehberger PA; Schulz N; Finberg KE; et al. (December 2003). "Localization and regulation of the ATP6V0A4 (a4) vacuolar H+
-ATPase subunit defective in an inherited form of distal renal tubular acidosis". J. Am. Soc. Nephrol. 14 (12): 3027–38. doi:10.1097/01.ASN.0000099375.74789.AB. PMID 14638902. - Stevens TH, Forgac M; Forgac (1997). "Structure, function and regulation of the vacuolar H+
-ATPase". Annu. Rev. Cell Dev. Biol. 13: 779–808. doi:10.1146/annurev.cellbio.13.1.779. PMID 9442887. - Stover EH; Borthwick KJ; Bavalia C; et al. (November 2002). "Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss". J. Med. Genet. 39 (11): 796–803. doi:10.1136/jmg.39.11.796. PMC 1735017. PMID 12414817.
- Sumner JP, Dow JA, Earley FG, Klein U, Jäger D, Wieczorek H; Dow; Earley; Klein; Jäger; Wieczorek (March 1995). "Regulation of plasma membrane V-ATPase activity by dissociation of peripheral subunits". J. Biol. Chem. 270 (10): 5649–53. doi:10.1074/jbc.270.10.5649. PMID 7890686.
- Susani L; Pangrazio A; Sobacchi C; et al. (September 2004). "TCIRG1-dependent recessive osteopetrosis: mutation analysis, functional identification of the splicing defects, and in vitro rescue by U1 snRNA". Hum. Mutat. 24 (3): 225–35. doi:10.1002/humu.20076. PMID 15300850.
- Whyteside G, Gibson L, Scott M, Finbow ME; Gibson; Scott; Finbow (June 2005). "Assembly of the yeast vacuolar H+
-ATPase and ATP hydrolysis occurs in the absence of subunit c". FEBS Lett. 579 (14): 2981–5. doi:10.1016/j.febslet.2005.04.049. PMID 15907326. - Zhang Z; Zheng Y; Mazon H; et al. (December 2008). "Structure of the yeast vacuolar ATPase". J. Biol. Chem. 283 (51): 35983–95. doi:10.1074/jbc.M805345200. PMC 2602884. PMID 18955482.
External links
- V-Type ATPase at the US National Library of Medicine Medical Subject Headings (MeSH)