Ehlers–Danlos syndrome
| Ehlers–Danlos syndrome | |
|---|---|
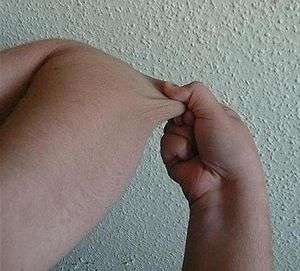 | |
| Individual with EDS displaying skin hyperelasticity | |
| Classification and external resources | |
| Specialty | Medical genetics |
| ICD-10 | Q79.6 (ILDS Q82.817) |
| ICD-9-CM | 756.83 |
| DiseasesDB | 4131 |
| MedlinePlus | 001468 |
| eMedicine | derm/696 ped/654 |
| Patient UK | Ehlers–Danlos syndrome |
| MeSH | D004535 |
| GeneReviews | |
| Orphanet | 98249 |
Ehlers–Danlos syndrome (EDS) is a group of genetic connective tissue disorders.[1] Symptoms can vary from mildly loose joints to life-threatening complications such as aortic dissection.[1] Chronic pain or early osteoarthritis may also occur.[2]
EDS is caused by a defect in the structure, production, or processing of collagen or proteins that interact with collagen. The collagen in connective tissue helps tissues resist deformation. Collagen is an important contributor to the physical strength of tissue; abnormal collagen renders these structures more elastic. In some cases, it can be life-threatening. People with joint pain may be misdiagnosed with hypochondriasis, depression, chronic fatigue syndrome, or other conditions.[2] There may be poor knowledge about EDS among practitioners.[3][4]
There is no cure for EDS. Treatment is supportive, including close monitoring of the digestive, excretory, and particularly the cardiovascular systems. Physical therapy, bracing, and corrective surgery may help with injuries and pain that tend to develop in certain types of EDS, although extra caution and special practices are advised to prevent permanent damage. EDS is a long term disease.[2]
EDS affects about 1 in 5,000 people globally.[1] Excess mobility was first described by Hippocrates in 400 BC.[5] The syndrome is named after two physicians, Edvard Ehlers from Denmark and Henri-Alexandre Danlos from France, who described it at the turn of the 20th century.[6][7]
Signs and symptoms

Signs vary widely based on which type of EDS the patient has. In each case, however, the signs are ultimately due to faulty or reduced amounts of collagen. EDS typically affects the joints, skin, and blood vessels. Following is a list of major signs and symptoms.
Musculoskeletal
- Hyper-flexible joints[7] (It is possible to be very flexible or have "double joints", but this is not the same as EDS.)
- Unstable joints that are prone to sprain, dislocation, subluxation, and hyperextension[8]
- Thoracic outlet syndrome
- Early onset of advanced osteoarthritis[9]
- Chronic degenerative joint disease[9]
- Swan neck deformity of the fingers[10]
- Boutonniere deformity of the fingers
- Tearing of tendons or muscles[11]
- Deformities of the spine, such as scoliosis (curvature of the spine), kyphosis (a thoracic hump), tethered spinal cord syndrome, and occipitoatlantoaxial hypermobility[12]
- Myalgia (muscle pain) and arthralgia (joint pain),[13] which may be severe
- Trendelenburg's sign[14]
- Osgood–Schlatter disease[15]
Skin
- Fragile skin that tears easily[9]
- Atrophic "cigarette paper" scars[1][16]
- Easy bruising[7]
- Redundant skin folds[9]
- Molluscoid pseudotumors,[17] especially on pressure points
- Subcutaneous spheroids[17]
- Livedo reticularis
- piezogenic papules [18]
Cardiovascular
- Arterial rupture[7]
- Valvular heart disease, such as mitral valve prolapse, which creates an increased risk for infective endocarditis during surgery. This may progress to a life-threatening degree.[19] Heart conduction abnormalities have been found in those with hypermobility form of EDS.[20]
- Dilation and/or rupture (aneurysm) of ascending aorta[21]
- Postural orthostatic tachycardia syndrome
- Raynaud's phenomenon
- Varicose veins
- Heart murmur
- Heart conduction abnormalities
Other manifestations or complications
- Hiatial hernia[17]
- Gastroesophageal reflux
- Gastrointestinal dysmotility[22]
- Dysautonomia[23]
- Gorlin's sign (touch tongue to nose) [24]
- Anal prolapse[17]
- Collapsed lung (spontaneous pneumothorax)[9]
- Nerve compression disorders (carpal tunnel syndrome, acroparesthesia, neuropathy, including Small fiber neuropathy)[25]
- Insensitivity to local anesthetics.[26]
- Arnold–Chiari malformation (brain disorder)[27]
- Platelet aggregation failure (platelets do not clump together properly)[28]
- Pregnancy complications: increased pain, mild to moderate peripartum bleeding, cervical insufficiency, uterine tearing,[11] or premature rupture of membranes.[29]
- Sleep apnea[30]
- Cranial vertebral instability: caused by trauma(s) to the head and neck areas such as concussion and whiplash. Ligaments in neck are unable to heal properly, therefore, the neck structure does not have the ability to support the skull, which can then sink into the brain stem blocking the normal flow of cerebral spinal fluid, leading to issues related to the autonomic nervous system failing to work properly.[31]
Because it is often undiagnosed or misdiagnosed in childhood, some instances of Ehlers–Danlos syndrome have been mischaracterized as child abuse.[32]
The pain associated with this condition is a serious complication.
Genetics

As mentioned under "Classification" above, only some variations of Ehlers-Danlos can be positively identified as tied to specific genetic variation.
Mutations in the following genes can cause subtypes of the Ehlers–Danlos syndrome:
- Fibrous proteins: COL1A1, COL1A2, COL3A1, COL5A1, COL5A2, and TNXB
- Enzymes: ADAMTS2, PLOD1, B4GALT7, DSE, and D4ST1/CHST14
Mutations in these genes usually alter the structure, production, or processing of collagen or proteins that interact with collagen. Collagen provides structure and strength to connective tissue. A defect in collagen can weaken connective tissue in the skin, bones, blood vessels, and organs, resulting in the features of the disorder.
Inheritance patterns depend on the type of Ehlers–Danlos syndrome. Most forms of the condition are inherited in an autosomal dominant pattern, which means only one of the two copies of the gene in question must be altered to cause the disorder. The minority are inherited in an autosomal recessive pattern, which means both copies of the gene must be altered for a person to be affected by the condition. It can also be an individual (de novo or "sporadic") mutation. Refer to the summary for each type of Ehlers–Danlos syndrome for a discussion of its inheritance pattern.
Diagnosis

A diagnosis can be made by an evaluation of medical history and clinical observation. The Brighton criteria is widely used to assess the degree of joint hypermobility. DNA and biochemical studies can help identify affected individuals. Diagnostic tests include collagen gene mutation testing, collagen typing via skin biopsy, echocardiogram, and lysyl hydroxylase or oxidase activity. However, these tests are not able to confirm all cases, especially in instances of an unmapped mutation, so clinical evaluation by a geneticist remains essential. If there are multiple affected individuals in a family, it may be possible to perform prenatal diagnosis using a DNA information technique known as a linkage study.
Classification
Up until 1997, the classification system for EDS included 10 specific types and also acknowledged that other extremely rare types existed. At this time, the classification system underwent an overhaul and was reduced to 6 major types using descriptive titles. Genetic specialists recognize that other types of this condition exist, but have only been documented in single families. Except for Hypermobility (type 3), some of the specific mutations involved have been identified and they can be precisely identified by genetic testing; this is valuable due to a great deal of variation in individual cases. However, negative genetic test results do not rule out the diagnosis, since not all of the mutations have been discovered; therefore the clinical presentation is very important.[33] Although the classifications are well defined, it is rare for a case to fit neatly in a single category, and cross-over symptoms lead to under-diagnosis or misdiagnosis. Therefore, patients should not rely on the "fact" of having a certain type of EDS if cross-over symptoms are evident because of possibly life-threatening symptoms. For example, it is possible for an individual with Classical EDS to exhibit symptoms of Hypermobility or Vascular EDS.
In decreasing order of prevalence in the population, the classifications are:
| Name | Number | Description | OMIM | Gene(s) |
| Hypermobility | type 3 | Affects 1 in 10,000 to 15,000 and is caused by an autosomal dominant or autosomal recessive mechanism. Mutations in either of two separate genes (which are also involved in Vascular EDS and Tenascin-X deficiency EDS, respectively) may lead to this variant. However, there has only been one report involving one family showing this mutation in COL3A1[34]), while there is some thought that the TNXB related EDS is actually its own separate subtype apart from type III. Joint hypermobility is the hallmark of this type, with less severe skin manifestations. Joint instability and chronic musculoskeletal pain are particularly prominent in this type. Patients with the Hypermobility Type experience frequent joint dislocations and subluxations (partial/incomplete dislocations), with or without trauma. As a result of dislocations and subluxations, pain is a common, severe, and a lifelong symptom of this type. Additionally, osteoarthritis is common, and many get it much earlier in life than expected.[35] Hypermobility type has also been associated with heart conduction abnormalities.[20]EDS type 3 is sometimes interchangeably called joints hypermobility syndrome (JHS). As no genetic test can identify or separate either conditions and because of the similarity of the diagnosis criteria and recommended treatments, many experts recommend they should be recognized as the same condition until further research is carried out.[36][37] | 130020 | TNXB, SometimesCOL3A1 (see description). Hypermobility type has also been associated with heart conduction abnormalities.[20] |
| Classical | types 1 & 2 | Affects approximately 1 in 20,000 to 50,000 people. It is caused by autosomal dominant mechanism and affects type-V collagen, as well as type-I collagen. Type 1 typically presents with severe skin involvement, and type 2 presents with mild to moderate skin involvement. Patients with the Classical Type may experience the same symptoms as the Hypermobility Type. The main difference between the Hypermobility and Classical Types is the Classical has more skin involvement while the Hypermobility Type has more joint involvement. Those with Classical EDS can also have severe joints issues like those with the Hypermobility Type. | 130000, 130010 | COL5A1, COL5A2, COL1A1 |
| Vascular | type 4 | Is an autosomal dominant defect in the type-III collagen synthesis; now thought to affect approximately 1 in 50,000 to 1 in 200,000.[38] Most are only diagnosed after rupturing, so it is believed that many more may well go undiagnosed. The vascular type is considered one of the more serious forms of Ehlers–Danlos syndrome because blood vessels and organs are fragile and prone to tearing (rupture), with a 60% increase of fatal rupture when infection is present. Many people with EDS type 4 express a characteristic, have a small stature with a slim build, and typically have thin, pale, translucent skin (veins can usually be seen on the chest, abdomen and other parts of body) with very easy bruising and propensity to develop ecchymoses (bruising without trauma). Degree of severity depends on the nature of the mutations involved. The current statistics, based largely on those only diagnosed after rupturing, indicate that about one in four people diagnosed with vascular type EDS develop a significant health problem by age 20 and more than 80 percent develop life-threatening complications by age 40. | 130050 | COL3A1 |
| Kyphoscoliosis | type 6 | Is an autosomal recessive defect due to deficiency of an enzyme called lysyl hydroxylase; it is very rare, with fewer than 60 cases reported. The kyphoscoliosis type is characterised by progressive curvature of the spine (scoliosis), thin conjunctiva with blue appearing sclera (eyes) and severe muscle weakness. | 225400, 229200 | PLOD1 |
| Arthrochalasia | types 7A & B | Is an autosomal dominant defect which is very rare, with roughly 30 cases reported. It affects type-I collagen. The arthrochalasia type was originally characterised by very loose joints and dislocations involving both hips at birth, but the diagnostic criteria has since been changed to allow diagnosis without the "hallmark" trait of hip dislocation at birth. Their joints are much looser than the Hypermobility Type. It is considered to be much more severe than Hypermobility Type. | 130060 | COL1A1, COL1A2 |
| Dermatosparaxis | type 7C | Autosomal recessive. Very rare, with approximately 10 cases reported. The dermatosparaxis type is characterized by extremely fragile and sagging skin. | 225410 | ADAMTS2 |
Other types
Forms of EDS in this category may present with soft, mildly stretchable skin, shortened bones, chronic diarrhea, joint hypermobility and dislocation, bladder rupture, or poor wound healing. Inheritance patterns in this group include X-linked recessive, autosomal dominant, and autosomal recessive. Examples of types of related syndromes other than those above reported in the medical literature include:
- 305200 – Type 5
- 130080 – Type 8 – unspecified gene, locus 12p13
- 225310 – Type 10 – unspecified gene, locus 2q34
- 608763 – Beasley–Cohen type
- 130070 – Progeroid form – B4GALT7
- 606408 – Due to Tenascin-X deficiency – TNXB
- 130090 – Type unspecified
- 601776 – D4ST1-Deficient Ehlers–Danlos syndrome (Adducted Thumb-Clubfoot Syndrome) CHST14
Differential diagnosis
There are several disorders that share some characteristics with Ehlers–Danlos syndrome. For example, in cutis laxa the skin is loose, hanging, and wrinkled. In EDS, the skin can be pulled away from the body but is elastic and returns to normal when let go. In Marfan syndrome, the joints are very mobile and similar cardiovascular complications occur. People with EDS tend to have a "Marfanoid" appearance (e.g., tall, skinny, long arms and legs, "spidery" fingers). However, physical appearance and features in several types of Ehlers-Danlos Syndrome also have characteristics including short stature, large eyes, and the appearance of a small mouth and chin, due to a small palate. The palate can have a high arch, causing dental crowding. Blood vessels can sometimes be easily seen through translucent skin, especially on the chest. In the past, Menkes disease, a copper metabolism disorder, was thought to be a form of Ehlers–Danlos syndrome. It is not uncommon for patients to be misdiagnosed with fibromyalgia, bleeding disorders or other disorders that can mimic EDS symptoms before a correct diagnosis is made. Because of these similar disorders and complications that can arise from an unmonitored case of EDS, a correct diagnosis is very important.[39] Pseudoxanthoma elasticum (PXE) is worth consideration in diagnosing a patient.
Management
There is no cure for Ehlers Danlos Syndrome. Treatment is palliative. Close monitoring of the cardiovascular system, physiotherapy, occupational therapy, and orthopedic instruments (e.g., wheelchairs, bracing, casting) may be helpful. Orthopedic instruments are helpful for the prevention of further joint damage, especially for long distances, although it is advised that individuals not become entirely dependent on them until there are no other options for mobility. One should avoid activities that cause the joint to lock or overextend.
A physician may prescribe casting to stabilize joints. Physicians may refer a patient to an orthotist for orthotic treatment (bracing). Physicians may also consult a physical and/or occupational therapist to help strengthen muscles and to teach people how to properly use and preserve their joints.[40][41]
There are different types of physiotherapy. Aquatic therapy promotes muscular development and coordination.[42] With manual therapy, the joint will be gently mobilized within the range of motion and/or manipulations.[40][41] If conservative therapy is not helpful, surgical repair of joints may be necessary. Medication to decrease pain or manage cardiac, digestive, or other related conditions may be prescribed. To decrease bruising and improve wound healing, some patients have responded to ascorbic acid (vitamin C).[43] Special precautions are often taken by medical care workers because of the sheer amount of complications that tend to arise in EDS patients. In Vascular EDS, signs of chest or abdominal pain are to be considered trauma situations.
In general, medical intervention is limited to symptomatic therapy. Before pregnancy, patients with EDS should have genetic counseling and familiarize themselves with the risks to their own bodies that pregnancy poses. Children with EDS should be provided with information about the disorder so they can understand why contact sports and other physically stressful activities should be avoided. Children should be taught early on that demonstrating the unusual positions they can maintain due to loose joints should not be done as this may cause early degeneration of the joints. Patients may find it hard to cope with the drawbacks of the disease. In this case, emotional support and behavioral and psychological therapy can be useful. Support groups can be immensely helpful for patients dealing with major lifestyle changes and poor health. Family members, teachers, and friends should be informed about EDS so they can accept and assist the child.
Surgery
The instability of joints, leading to (sub)luxations and joint pain, often require surgical intervention in patients with Ehlers–Danlos syndrome. Instability of almost all joints can happen but appear most often in the lower and upper extremities, with the wrist, fingers, shoulder, knee, hip, and ankle being most common.[40]
Common surgical procedures are joint debridement, tendon replacements, capsulorraphy, and arthroplasty. Studies have shown that after surgery, degree of stabilization, pain reduction, and patient satisfaction can improve, but surgery does not guarantee an optimal result: Patients and surgeons report being dissatisfied with the results. Consensus is that conservative treatment is more effective than surgery,[20] particularly since patients have extra risks of surgical complications due to the disease. Three basic surgical problems arise due to EDS: the strength of the tissues is decreased, which makes the tissue less suitable for surgery; the fragility of the blood vessels can cause problems during surgery; and wound healing is often delayed or incomplete.[40] If considering surgical intervention, it would be prudent to seek care from a surgeon with extensive knowledge and experience in treating patients with EDS and joint hypermobility issues.
Studies have shown that local anesthetics, arterial catheters and central venous catheters cause a higher risk in haematoma formation in patients with Ehlers–Danlos syndrome. Ehlers-Danlos patients also show a resistance to local anaesthetics.[44] Resistance to Xylocaine and Bupivacaine is not uncommon, and Carbocaine tends to work better in EDS patents. Special recommendations for anesthesia in EDS patients are prepared by orphan anesthesia.eu and deal with all aspects of anesthesia for EDS patients.[45] Detailed recommendations for anesthesia and perioperative care of patients with EDS should be used to improve patient safety.[46]
Surgery with Ehlers-Danlos patients requires careful tissue handling and a longer immobilization afterward.
Prognosis
The outlook for individuals with EDS depends on the type of EDS they have. Symptoms vary in severity, even within one sub-type, and the frequency of complications changes individually. Some people have negligible symptoms while others are severely restricted in their daily life. Extreme joint instability, chronic musculoskeletal pain, degenerative joint disease, frequent injuries, and spinal deformities may limit mobility. Severe spinal deformities may affect breathing. In the case of extreme joint instability, dislocations may result from simple tasks such as rolling over in bed or turning a doorknob. Secondary conditions such as autonomic dysfunction or cardiovascular problems, occurring in any type, can affect prognosis and quality of life. Severe mobility-related disability is seen more often in Hypermobility-type than in Classical-type or Vascular-type.
Although all types are potentially life-threatening, the majority of individuals will have a normal lifespan. However, those with blood vessel fragility have a high risk of fatal complications. Arterial rupture is the most common cause of sudden death in EDS. Spontaneous arterial rupture most often occurs in the second or third decade, but can occur at any time. The median life-expectancy in the population with Vascular EDS is 48 years.[47]
EDS is a lifelong condition. Affected individuals may face social obstacles related to their disease daily. Some people with EDS have reported living with fear of significant and painful ruptures, their condition worsening, becoming unemployed due to physical and emotional burdens, and social stigmatization in general.
Epidemiology
Ehlers–Danlos syndrome is an inherited disorder estimated to occur in about 1 in 5,000 births worldwide. Initially, prevalence estimates ranged from 1 in 250,000 to 1 in 500,000 people, but these estimates were soon found to be vastly inaccurate as the disorder received further study and medical professionals became more adept at accurately diagnosing EDS. In fact, many experts now believe that Ehlers–Danlos syndrome may be far more common than the currently accepted estimate due to the wide range of severities with which the disorder presents.[48] The prevalence of the six types differs dramatically. The most commonly occurring is the Hypermobility type, followed by the Classical type. The other types of Ehlers–Danlos syndrome are very rare. For example, fewer than 10 infants and children with the dermatosparaxis type have been described worldwide. Ehlers–Danlos affects males and females of all racial and ethnic backgrounds, although some types are more common among certain groups than others.
Society and culture

- In the 19th century, there were several sideshow performers billed as The Elastic Skin Man, The India Rubber Man and Frog Boy. They included such well-known individuals (in their time) as Felix Wehrle, James Morris and Avery Childs.[49]
- Several current celebrities have EDS:
- Actress Cherylee Houston has type 3 EDS and uses a wheelchair; she made history by becoming Coronation Street's first full-time disabled actress.[50]
- The condition may have contributed to the virtuoso violinist Niccolò Paganini's skill as he was able to play wider fingerings than the normal violinist.[51]
- Martina Stawski from the YouTube channel Eatyourkimchi has type 3 EDS and briefly addresses the limitations of her condition on some v-log formatted videos on the channel.
- Eric Shaun Lynch, aka Eric the Midget and later Eric the Actor, a member of Howard Stern's Wack Pack had Ehlers–Danlos Syndrome Type VII.
- Science blogger Yvette d'Entremont aka Science Babe has Ehlers–Danlos Syndrome.[52]
- Adult film star Mandy Morbid has discussed the impact EDS has on her mobility and her life.[53]
- Rei Haycraft, lead singer for the hard rock band Raimee, artist, and illustrator. Haycraft has created songs and a documentary about living with Ehlers-Danlos, as well as written songs about its impact on her life.[54][55][56]
- The condition is mentioned in the song "Dorsal Horn Concerto" by the British comedy band the Amateur Transplants.[57]
- The condition has been mentioned in many television shows, including:
- The subject of the Season 7 premiere of the A&E series Intervention, Linda, claims to have EDS.[58]
Other animals

Ehlers–Danlos-like syndromes have been shown to be hereditary in Himalayan cats, some domestic shorthair cats, and in certain breeds of cattle. It is seen as a sporadic condition in domestic dogs.
Degenerative suspensory ligament desmitis (DSLD) is a similar condition seen in many breeds of horses. It was originally notated in the Peruvian Paso and thought to be a condition of overwork and older age. However, the disease is being recognized in all age groups and all activity levels. It has even been noted in newborn foals. The latest research has led to the renaming of the disease as equine systemic proteoglycan accumulation, after the possible systemic and hereditary components being delineated by the University of Georgia.[59][60]
References
- 1 2 3 4 "Ehlers-Danlos syndrome". Genetics Home Reference. Retrieved 8 May 2016.
- 1 2 3 Lawrence, Elizabeth J. (2005). "The clinical presentation of Ehlers-Danlos syndrome". Advances in Neonatal Care. 5 (6): 301–14. doi:10.1016/j.adnc.2005.09.006. PMID 16338669.
- ↑ Ross, J.; Grahame, R. (2011). "Joint hypermobility syndrome". BMJ. 342: c7167. doi:10.1136/bmj.c7167. PMID 21252103.
- ↑ Castori, Marco (2012). "Ehlers-Danlos Syndrome, Hypermobility Type: An Underdiagnosed Hereditary Connective Tissue Disorder with Mucocutaneous, Articular, and Systemic Manifestations". ISRN Dermatology. 2012: 751768. doi:10.5402/2012/751768. PMC 3512326
 . PMID 23227356.
. PMID 23227356. - ↑ Beighton, Peter H.; Grahame, Rodney; Bird, Howard A. (2011). Hypermobility of Joints. Springer. p. 1. ISBN 9781848820852.
- ↑ "Uncovered: How U.S. Health System Can Fail Even the Insured – A Woman Endures 16-Month Odyssey To Get a Diagnosis", John Carreyrou, Wall Street Journal, November 16, 2007
- 1 2 3 4 Byers PH, Murray ML (2012). "Heritable collagen disorders: the paradigm of Ehlers-Danlos syndrome". Journal of Investigative Dermatology. 132 (E1): E6–11. doi:10.1038/skinbio.2012.3. PMID 23154631.
- ↑ Lawrence EJ (2005). "The clinical presentation of Ehlers–Danlos syndrome". Adv Neonatal Care. 5 (6): 301–14. doi:10.1016/j.adnc.2005.09.006. PMID 16338669.
- 1 2 3 4 5 "Ehlers–Danlos Syndrome". Mayo Clinic. Retrieved 25 May 2012.
- ↑ Erçöçen AR, Yenidünya MO, Yilmaz S, Ozbek MR (1997). "Dynamic swan neck deformity in a patient with Ehlers-Danlos syndrome". J Hand Surg Br. 22 (1): 128–30. doi:10.1016/S0266-7681(97)80039-3. PMID 9061548.
- 1 2 "Vascular Type-EDS". Ehlers–Danlos Syndrome Network C.A.R.E.S. Inc.
- ↑ Milhorat TH, Bolognese PA, Nishikawa M, McDonnell NB, Francomano CA (December 2007). "Syndrome of occipitoatlantoaxial hypermobility, cranial settling, and chiari malformation type I in patients with hereditary disorders of connective tissue". Journal of Neurosurgery. Spine. 7 (6): 601–9. doi:10.3171/SPI-07/12/601. PMID 18074684.
- ↑ Gedalia A, Press J, Klein M, Buskila D (1993). "Joint hypermobility and fibromyalgia in schoolchildren". Annals of the Rheumatic Diseases. 52 (7): 494–6. doi:10.1136/ard.52.7.494. PMC 1005086. PMID 8346976.
- ↑ Dommerholt, Jan. "CSF Ehlers Danlos Colloquium, Dr Jan Dommerholt". Chiari & Syringomyelia Foundation. Retrieved 10 June 2013.
- ↑ Vigorita, Vincent J; Mintz, Douglas; Ghelman, Bernard (2008). Orthopaedic pathology (2nd ed.). Philadelphia: Lippincott Williams and Wilkins. pp. 5–6. ISBN 0781796709.
- ↑ "Unusual scars in a young female patient". Postgraduate Medical Journal. 81 (957): e5–e5. 2005-07-01. ISSN 1469-0756.
- 1 2 3 4 "Classical Type-EDS". Ehlers–Danlos Syndrome Network C.A.R.E.S Inc.
- ↑ https://www.dermnetnz.org/topics/piezogenic-papules
- ↑ Pagon RA, Adam MP, Ardinger HH, et al. (1993). "Ehlers-Danlos Syndrome, Hypermobility Type". PMID 20301456.
- 1 2 3 4 Camerota F, Castori M, Celletti C, Colotto M, Amato S, Colella A, Curione M, Danese C (2014). "Heart rate, conduction and ultrasound abnormalities in adults with joint hypermobility syndrome/Ehlers-Danlos syndrome, hypermobility type". Clin. Rheumatol. 33 (7): 981–7. doi:10.1007/s10067-014-2618-y. PMID 24752348.
- ↑ Wenstrup, R.J; et al. (2001). The Ehlers–Danlos Syndromes: Management of Genetic Syndromes. pp. 131–149.
- ↑ Brockway, Laura (April 2016). "Gastrointestinal manifestations of Ehlers-Danlos syndrome (hypermobility type)". Ehlers-Danlos Support UK.
- ↑ "Ehlers-Danlos Syndrome". Underlying Causes of Dysautonomia. Dysautonomia International. 2012.
- ↑ Létourneau Y, Pérusse R, Buithieu H (2001). "Oral manifestations of Ehlers-Danlos syndrome". J Can Dent Assoc. 67 (6): 330–4. PMID 11450296.
- ↑ "Ehlers–Danlos syndrome: Definition from". Answers.com. Retrieved 2014-02-27.
- ↑ Arendt-Nielsen, Lars. "Patients Suffering from Ehlers Danlos Syndrome Type III Do Not Respond to Local Anesthetics".
- ↑ Neurosurgery spine Archived October 19, 2013, at the Wayback Machine.
- ↑ MedlinePlus Encyclopedia Ehlers-Danlos syndrome
- ↑ Lind J, Wallenburg HC (April 2002). "Pregnancy and the Ehlers–Danlos syndrome: a retrospective study in a Dutch population". Acta Obstetricia et Gynecologica Scandinavica. 81 (4): 293–300. doi:10.1034/j.1600-0412.2002.810403.x. PMID 11952457.
- ↑ Guilleminault C, Primeau M, Chiu HY, Yuen KM, Leger D, Metlaine A (2013). "Sleep-disordered breathing in Ehlers-Danlos syndrome: a genetic model of OSA". Chest. 144 (5): 1503–11. doi:10.1378/chest.13-0174. PMID 23929538.
- ↑ Henderson, Fraser (2015). "Indices of Cranio-vertebral Instability". Funded Research. Chiari & Syringomyelia Foundation.
- ↑ The Press Enterprise, Redlands mother stung by untrue suspicions presses for accountability in child abuse inquiries, 2008-04-03
- ↑ "What is EDS | The Ehlers-Danlos National Foundation". www.ednf.org. Retrieved 2016-01-06.
- ↑ Narcisi P, Richards AJ, Ferguson SD, Pope FM (1994). "A family with Ehlers-Danlos syndrome type III/articular hypermobility syndrome has a glycine 637 to serine substitution in type III collagen". Hum. Mol. Genet. 3 (9): 1617–20. doi:10.1093/hmg/3.9.1617. PMID 7833919.
- ↑ Levy 2016
- ↑ "Ehlers Danlos UK - JHS vs EDS". www.ehlers-danlos.org. Retrieved 2016-12-01.
- ↑ "Hypermobility Syndromes Association » JHS v EDS Hypermobility- Same Thing?". hypermobility.org. Retrieved 2016-12-01.
- ↑ Pepin, Murray & Byers 2015
- ↑ "Ehlers-Danlos Syndrome". Rarediseases.about.com. 2006-05-25. Retrieved 2014-02-27.
- 1 2 3 4 Rombaut L, Malfait F, De Wandele I, Cools A, Thijs Y, De Paepe A, Calders P (July 2011). "Medication, Surgery, and Physiotherapy Among Patients With the Hypermobility Type of Ehlers-Danlos Syndrome". Archives of physical medicine and rehabilitation. 92 (7): 1106–12. doi:10.1016/j.apmr.2011.01.016. PMID 21636074.
- 1 2 Woerdeman LA, Ritt MJ, Meijer B, Maas M (2000). "Wrist problems in patients with Ehlers-Danlos syndrome". European Journal of Plastic Surgery. 23 (4): 208–210. doi:10.1007/s002380050252.
- ↑ Callewaert B, Malfait F, Loeys B, De Paepe A (March 2008). "Ehlers-Danlos syndromes and Marfan syndrome". Best practice & research. Clinical Rheumatology. 22 (1): 165–189. doi:10.1016/j.berh.2007.12.005. PMID 18328988.
- ↑ Genetics of Ehlers-Danlos Syndrome~treatment at eMedicine
- ↑ Parapia, Liakat A.; Jackson, Carolyn (2008). "Ehlers-Danlos syndrome – a historical review". British Journal of Haematology. 141 (1): 32–5. doi:10.1111/j.1365-2141.2008.06994.x. PMID 18324963.
- ↑ OrphanAnesthesia Guidelines
- ↑ Wiesmann, Thomas; Castori, Marco; Malfait, Fransiska; Wulf, Hinnerk (2014). "Recommendations for anesthesia and perioperative management in patients with Ehlers-Danlos syndrome(s)". Orphanet Journal of Rare Diseases. 9: 109. doi:10.1186/s13023-014-0109-5. PMC 4223622. PMID 25053156.
- ↑ Pepin, Melanie; Schwarze, Ulrike; Superti-Furga, Andrea; Byers, Peter H. (2000). "Clinical and Genetic Features of Ehlers–Danlos Syndrome Type IV, the Vascular Type". New England Journal of Medicine. 342 (10): 673–80. doi:10.1056/NEJM200003093421001. PMID 10706896.
- ↑ "Ehlers-Danlos Syndrome: Epidemiology". Medscape.com. Retrieved 2014-02-27.
- ↑ Mitchell, Michael (1979). Monsters of the Gilded Age: The photographs of Charles Eisenmann. Toronto: Gage. pp. 99, 100. ISBN 0771595212. OCLC 6892718.
- ↑ "Houston hits out at "preconceived ideas" - Coronation Street News - Soaps". Digital Spy. 2010-05-22. Retrieved 2014-02-27.
- ↑ Yücel D (1995). "Was Paganini born with Ehlers-Danlos syndrome phenotype 4 or 3?". Clin. Chem. 41 (1): 124–5. PMID 7813066.
- ↑ "You Can Dislocate a Rib By Sneezing; My Life With Ehlers Danlos Syndrome". SciBabe. 1 July 2016.
- ↑ Kane, Kimberly (16 October 2012). "Zak Loves Mandy, And They'll Keep Making Art and Porn Until One of Them Dies". Vice.
- ↑ "But You Don't Look Sick: Living With Chronic Pain (Ehlers Danlos Syndrome)". YouTube. Retrieved 2014-02-27.
- ↑ "'I can't have sex without dislocating my hip' says student, 19". Mail Online. Retrieved 2016-02-25.
- ↑ Barry, Aoife. ""People are being left to rot": Rare disease sufferers feel let down by health service". TheJournal.ie. Retrieved 2016-02-25.
- ↑ Video on YouTube
- ↑ "Intervention – Episode Guide". www.aetv.com. Retrieved 24 November 2012.
- ↑ Halper, Jaroslava; Kim, Byoungjae; Khan, Ahrar; Yoon, Jung; Mueller, PO (2006). "Degenerative suspensory ligament desmitis as a systemic disorder characterized by proteoglycan accumulation". BMC Veterinary Research. 2: 12. doi:10.1186/1746-6148-2-12. PMC 1459153. PMID 16611357.
- ↑ Kim, Byoungjae; Yoon, Jung Hae; Zhang, Jian; Eric Mueller, P.O.; Halper, Jaroslava (2010). "Glycan profiling of a defect in decorin glycosylation in equine systemic proteoglycan accumulation, a potential model of progeroid form of Ehlers-Danlos syndrome". Archives of Biochemistry and Biophysics. 501 (2): 221–31. doi:10.1016/j.abb.2010.06.017. PMID 20599673.
External links
| Wikimedia Commons has media related to Ehlers-Danlos syndrome. |
- Ehlers–Danlos syndrome at DMOZ
- "Ehlers-Danlos syndrome". University of Washington Medicine, Orthopaedics and Sports Medicine. Multi-page explanation of EDS, including symptoms, genetics, diagnosis, and treatment
- "Ehlers-Danlos syndrome". Genetics Home Reference. US National Library of Medicine. 8 November 2016.
- Pagon RA, Bird TD, Dolan CR; et al., eds. (1993–2016). GeneReviews™ [Internet]. Seattle WA: University of Washington, Seattle. ISSN 2372-0697.
- Malfait, Fransiska; Wenstrup, Richard; De Paepe, Anne (18 August 2011). Ehlers-Danlos Syndrome, Classic Type. PMID 20301422. NBK1244.
- Pepin, Melanie G; Byers, Mitzi L; Murray, Peter H (19 November 2015). Vascular Ehlers-Danlos Syndrome. PMID 20301667. NBK1494.
- Levy, Howard P (31 March 2016). Ehlers-Danlos Syndrome, Hypermobility Type. PMID 20301456. NBK1279.
- Yeowell, Heather N; Steinmann, Beat (24 January 2013). Ehlers-Danlos Syndrome, Kyphoscoliotic Form. PMID 20301635. NBK1462.
- Castori M, Morlino S, Celletti C, Celli M, Morrone A, Colombi M, Camerota F, Grammatico P (2012). "Management of pain and fatigue in the joint hypermobility syndrome (a.k.a. Ehlers-Danlos syndrome, hypermobility type): principles and proposal for a multidisciplinary approach". Am. J. Med. Genet. A. 158A (8): 2055–70. doi:10.1002/ajmg.a.35483. PMID 22786715.
- "Ehlers–Danlos syndrome". Published Guidelines: OrphanAnesthesia. German Society of Anesthesiology and Intensive Care Medicine.