Virtual karyotype
Virtual karyotype is the digital information reflecting a karyotype, resulting from the analysis of short sequences of DNA from specific loci all over the genome, which are isolated and enumerated.[1] It detects genomic copy number variations at a higher resolution level than conventional karyotyping or chromosome-based comparative genomic hybridization (CGH).[2] The main methods used for creating virtual karyotypes are array-comparative genomic hybridization and SNP arrays.
Background
A karyotype (Fig 1) is the characteristic chromosome complement of a eukaryote species.[3][4] A karyotype is typically presented as an image of the chromosomes from a single cell arranged from largest (chromosome 1) to smallest (chromosome 22), with the sex chromosomes (X and Y) shown last. Historically, karyotypes have been obtained by staining cells after they have been chemically arrested during cell division. Karyotypes have been used for several decades to identify chromosomal abnormalities in both germline and cancer cells. Conventional karyotypes can assess the entire genome for changes in chromosome structure and number, but the resolution is relatively coarse, with a detection limit of 5-10Mb.

Method
Recently, platforms for generating high-resolution karyotypes in silico from disrupted DNA have emerged, such as array comparative genomic hybridization (arrayCGH) and SNP arrays. Conceptually, the arrays are composed of hundreds to millions of probes which are complementary to a region of interest in the genome. The disrupted DNA from the test sample is fragmented, labeled, and hybridized to the array. The hybridization signal intensities for each probe are used by specialized software to generate a log2ratio of test/normal for each probe on the array. Knowing the address of each probe on the array and the address of each probe in the genome, the software lines up the probes in chromosomal order and reconstructs the genome in silico (Fig 2 and 3).
Virtual karyotypes have dramatically higher resolution than conventional cytogenetics. The actual resolution will depend on the density of probes on the array. Currently, the Affymetrix SNP6.0 is the highest density commercially available array for virtual karyotyping applications. It contains 1.8 million polymorphic and non-polymorphic markers for a practical resolution of 10-20kb—about the size of a gene. This is approximately 1000-fold greater resolution than karyotypes obtained from conventional cytogenetics.
Virtual karyotypes can be performed on germline samples for constitutional disorders,[5][6] and clinical testing is available from dozens of CLIA certified laboratories (genetests.org). Virtual karyotyping can also be done on fresh or formalin-fixed paraffin-embedded tumors.[7][8][9] CLIA-certified laboratories offering testing on tumors include Creighton Medical Laboratories (fresh and paraffin embedded tumor samples) and CombiMatrix Molecular Diagnostics (fresh tumor samples).


Different platforms for virtual karyotyping
Array-based karyotyping can be done with several different platforms, both laboratory-developed and commercial. The arrays themselves can be genome-wide (probes distributed over the entire genome) or targeted (probes for genomic regions known to be involved in a specific disease) or a combination of both. Further, arrays used for karyotyping may use non-polymorphic probes, polymorphic probes (i.e., SNP-containing), or a combination of both. Non-polymorphic probes can provide only copy number information, while SNP arrays can provide both copy number and loss-of-heterozygosity (LOH) status in one assay. The probe types used for non-polymorphic arrays include cDNA, BAC clones (e.g., BlueGnome), and oligonucleotides (e.g., Agilent, Santa Clara, CA, USA or Nimblegen, Madison, WI, USA). Commercially available oligonucleotide SNP arrays can be solid phase (Affymetrix, Santa Clara, CA, USA) or bead-based (Illumina, SanDiego, CA, USA). Despite the diversity of platforms, ultimately they all use genomic DNA from disrupted cells to recreate a high resolution karyotype in silico. The end product does not yet have a consistent name, and has been called virtual karyotyping,[8][10] digital karyotyping,[11] molecular allelokaryotyping,[12] and molecular karyotyping.[13] Other terms used to describe the arrays used for karyotyping include SOMA (SNP oligonucleotide microarrays)[14] and CMA (chromosome microarray).[15][16] Some consider all platforms to be a type of array comparative genomic hybridization (arrayCGH), while others reserve that term for two-dye methods, and still others segregate SNP arrays because they generate more and different information than two-dye arrayCGH methods.
Applications
Detecting copy-number changes
Copy number changes can be seen in both germline and tumor samples. Copy number changes can be detected by arrays with non-polymorphic probes, such as arrayCGH, and by SNP-based arrays. Human beings are diploid, so a normal copy number is always two for the non-sex chromosomes.
- Deletions: A deletion is the loss of genetic material. The deletion can be heterozygous (copy number of 1) or homozygous (copy number of 0, nullisomy). Microdeletion syndromes are examples of constitutional disorders due to small deletions in germline DNA. Deletions in tumor cells may represent the inactivation of a tumor suppressor gene, and may have diagnostic, prognostic, or therapeutic implications.
- Gains: A copy number gain represents the gain of genetic material. If the gain is of just one additional copy of a segment of DNA, it may be called a duplication (Fig 4). If there is one extra copy of an entire chromosome, it may be called a trisomy. Copy number gains in germline samples may be disease-associated or may be a benign copy number variant. When seen in tumor cells, they may have diagnostic, prognostic, or therapeutic implications.
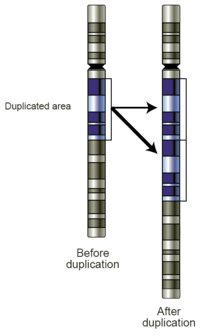
- Amplifications: Technically, an amplification is a type of copy number gain in which there is a copy number >10. In the context of cancer biology, amplifications are often seen in oncogenes. This could indicate a worse prognosis, help categorize the tumor, or indicate drug eligibility. An example of drug eligibility is Her2Neu amplification and Herceptin, and an image of Her2Neu amplification detected by SNP array virtual karyotyping is provided (Fig 5).
 Fig 5. Her2 Amplification by SNP array virtual karyotype.
Fig 5. Her2 Amplification by SNP array virtual karyotype.
Loss of heterozygosity (LOH), autozygous segments, and uniparental disomy
Autozygous segments and uniparental disomy (UPD) are diploid/'copy neutral' genetic findings and therefore are only detectable by SNP-based arrays. Both autozygous segments and UPD will show loss of heterozygosity (LOH) with a copy number of two by SNP array karyotyping. The term Runs of Homozgygosity (ROH), is a generic term that can be used for either autozygous segments or UPD.
- Autozygous segment: An autozygous segment is bi-parental and seen only in the germline. They are extended runs of homozygous markers in the genome, and they occur when an identical haplotype block is inherited from both parents. They are also called "identical by descent" (IBD) segments, and they can be used for homozygosity mapping.[17][18]
- Uniparental Disomy: UPD occurs when both copies of a gene or genomic region are inherited from the same parent. This is uniparental, in contrast to autozygous segments which are bi-parental. When present in the germline, they can be harmless or associated with disease, such as Prader-Willi or Angelman syndromes. Also in contrast to autozygosity, UPD can develop in tumor cells, and this is referred to as acquired UPD or copy neutral LOH in the literature (Fig 6). Acquired UPD is quite common in both hematologic and solid tumors, and is reported to constitute 20 to 80% of the LOH seen in human tumors.[19][20][21][22] Acquired UPD can serve as the 2nd hit in the Knudson Two Hit Hypothesis of Tumorigenesis, and thus can be the biological equivalent of a deletion.[23] Because this type of lesion cannot be detected by arrayCGH, FISH, or conventional cytogenetics, SNP-based arrays are preferred for virtual karyotyping of tumors.
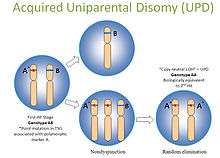 Fig 6. Copy neutral LOH/uniparental disomy
Fig 6. Copy neutral LOH/uniparental disomy

Figure 7 is a SNP array virtual karyotype from a colorectal carcinoma demonstrating deletions, gains, amplifications, and acquired UPD (copy neutral LOH).
Examples of clinical cancer applications
A virtual karyotype can be generated from nearly any tumor, but the clinical meaning of the genomic aberrations identified are different for each tumor type. Clinical utility varies and appropriateness is best determined by an oncologist or pathologist in consultation with the laboratory director of the lab performing the virtual karyotype. Below are examples of types of cancers where the clinical implications of specific genomic aberrations are well established. This list is representative, not exhaustive. The web site for the Cytogenetics Laboratory at Wisconsin State Laboratory of Hygiene has additional examples of clinically relevant genetic changes that are readily detectable by virtual karyotyping.
Neuroblastoma
Based on a series of 493 neuroblastoma samples, it has been reported that overall genomic pattern, as tested by array-based karyotyping, is a predictor of outcome in neuroblastoma:[24]
- Tumors presenting exclusively with whole chromosome copy number changes were associated with excellent survival.
- Tumors presenting with any kind of segmental chromosome copy number changes were associated with a high risk of relapse.
- Within tumors showing segmental alterations, additional independent predictors of decreased overall survival were MYCN amplification, 1p and 11q deletions, and 1q gain.
Earlier publications categorized neuroblastomas into three major subtypes based on cytogenetic profiles:[25]
- Subtype 1: favorable neuroblastoma with near triploidy and a predominance of numerical gains and losses, mostly representing non-metastatic NB stages 1, 2 and 4S.
- Subtypes 2A and 2B: found in unfavorable widespread neuroblastoma, stages 3 and 4, with 11q loss and 17q gain without MYCN amplification (subtype 2A) or with MYCN amplification often together with 1p deletions and 17q gain (subtype 2B).
Wilms' tumor
Tumor-specific loss-of-heterozygosity (LOH) for chromosomes 1p and 16q identifies a subset of Wilms' tumor patients who have a significantly increased risk of relapse and death. LOH for these chromosomal regions can now be used as an independent prognostic factor together with disease stage to target intensity of treatment to risk of treatment failure.[26][27]
Renal-cell carcinoma
Renal epithelial neoplasms have characteristic cytogenetic aberrations that can aid in classification.[28] See also Atlas of Genetics and Cytogenetics in Oncology and Haematology.
- Clear cell carcinoma: loss of 3p
- Papillary carcinoma: trisomy 7 and 17
- Chromophobe carcinoma: hypodiploid with loss of chromosomes 1, 2, 6, 10, 13, 17, 21
Array-based karyotyping can be used to identify characteristic chromosomal aberrations in renal tumors with challenging morphology.[8][10] Array-based karyotyping performs well on paraffin embedded tumors[29] and is amenable to routine clinical use.
In addition, recent literature indicates that certain chromosomal aberrations are associated with outcome in specific subtypes of renal epithelial tumors.[30]
Clear cell renal carcinoma: del 9p and del 14q are poor prognostic indicators.[31][32]
Papillary renal cell carcinoma: duplication of 1q marks fatal progression.[33]
Chronic lymphocytic leukemia
Array-based karyotyping is a cost-effective alternative to FISH for detecting chromosomal abnormalities in chronic lymphocytic leukemia (CLL). Several clinical validation studies have shown >95% concordance with the standard CLL FISH panel.[12][34][35][36][37] In addition, many studies using array-based karyotyping have identified 'atypical deletions' missed by the standard FISH probes and acquired uniparental disomy at key loci for prognostic risk in CLL.[38][39]
Four main genetic aberrations are recognized in CLL cells that have a major impact on disease behavior.[40]
- Deletions of part of the short arm of chromosome 17 (del 17p) which target p53 are particularly deleterious. Patients with this abnormality have significantly short interval before they require therapy and a shorter survival. This abnormality is found in 5–10% of patients with CLL.
- Deletions of the long arm on chromosome 11 (del 11q) are also unfavorable although not to the degree seen with del 17p. The abnormality targets the ATM gene and occurs infrequently in CLL (5–10%).
- Trisomy 12, an additional chromosome 12, is a relatively frequent finding occurring in 20–25% of patients and imparts an intermediate prognosis.
- Deletion of 13q14 (del 13q14) is the most common abnormality in CLL with roughly 50% of patients with cells containing this defect. When del 13q14 is seen in isolation, patients have the best prognosis and most will live many years, even decades, without the need for therapy.
Multiple myeloma
Avet-Loiseau, et al. in Journal of Clinical Oncology, used SNP array karyotyping of 192 multiple myeloma (MM) samples to identify genetic lesions associated with prognosis, which were then validated in a separate cohort (n = 273).[41] In MM, lack of a proliferative clone makes conventional cytogenetics informative in only ~30% of cases. FISH panels are useful in MM, but standard panels would not detect several key genetic abnormalities reported in this study.
- Virtual karyotyping identified chromosomal abnormalities in 98% of MM cases
- del(12p13.31)is an independent adverse marker
- amp(5q31.1) is a favorable marker
- The prognostic impact of amp(5q31.1) over-rides that of hyperdiploidy and also identifies patients who greatly benefit from high-dose therapy.
Array-based karytyping cannot detect balanced translocations, such as t(4;14) seen in ~15% of MM. Therefore, FISH for this translocation should also be performed if using SNP arrays to detect genome-wide copy number alterations of prognostic significance in MM.
Medulloblastoma
Array-based karyotyping of 260 medulloblastomas by Pfister S, et al. resulted in the following clinical subgroups based on cytogenetic profiles:[42]
- Poor prognosis: gain of 6q or amplification of MYC or MYCN
- Intermediate: gain of 17q or an i(17q) without gain of 6q or amplification of MYC or MYCN
- Excellent prognosis: 6q and 17q balanced or 6q deletion
Oligodendroglioma
The 1p/19q co-deletion is considered a "genetic signature" of oligodendroglioma. Allelic losses on 1p and 19q, either separately or combined, are more common in classic oligodendrogliomas than in either astrocytomas or oligoastrocytomas.[43] In one study, classic oligodendrogliomas showed 1p loss in 35 of 42 (83%) cases, 19q loss in 28 of 39 (72%), and these were combined in 27 of 39 (69%) cases; there was no significant difference in 1p/19q loss of heterozygosity status between low-grade and anaplastic oligodendrogliomas.[43] 1p/19q co-deletion has been correlated with both chemosensitivity and improved prognosis in oligodendrogliomas.[44][45] Most larger cancer treatment centers routinely check for the deletion of 1p/19q as part of the pathology report for oligodendrogliomas. The status of the 1p/19q loci can be detected by FISH or virtual karyotyping. Virtual karyotyping has the advantage of assessing the entire genome in one assay, as well as the 1p/19q loci. This allows assessment of other key loci in glial tumors, such as EGFR and TP53 copy number status.
Whereas the prognostic relevance of 1p and 19q deletions is well established for anaplastic oligodendrogliomas and mixed oligoastrocytomas, the prognostic relevance of the deletions for low-grade gliomas is more controversial. In terms of low-grade gliomas, a recent study also suggests that 1p/19q co-deletion may be associated with a (1;19)(q10;p10) translocation which, like the combined 1p/19q deletion, is associated with superior overall survival and progression-free survival in low-grade glioma patients.[46] Oligodendrogliomas show only rarely mutations in the p53 gene, which is in contrast to other gliomas.[47] Epidermal growth factor receptor amplification and whole 1p/19q codeletion are mutually exclusive and predictive of completely different outcomes, with EGFR amplification predicting poor prognosis.[48]
Glioblastoma
Yin et al.[49] studied 55 glioblastoma and 6 GBM cell lines using SNP array karyotyping. Acquired UPD was identified at 17p in 13/61 cases. A significantly shortened survival time was found in patients with 13q14 (RB) deletion or 17p13.1 (p53) deletion/acquired UPD. Taken together, these results suggest that this technique is a rapid, robust, and inexpensive method to profile genome-wide abnormalities in GBM. Because SNP array karyotyping can be performed on paraffin embedded tumors, it is an attractive option when tumor cells fail to grow in culture for metaphase cytogenetics or when the desire for karyotyping arises after the specimen has been formalin fixed.
The importance of detecting acquired UPD (copy neutral LOH) in glioblastoma:
- Of patients with 17p abnormality, ~50% were deletions and ~50% were aUPD
- Both 17p del and 17p UPD were associated with worse outcome.
- 9/13 had homozygous TP53 mutations underlying the 17p UPD.
In addition, in cases with uncertain grade by morphology, genomic profiling can assist in diagnosis.
- Concomitant gain of 7 and loss of 10 is essentially pathognomonic for GBM[50]
- EGFR amplification, loss of PTEN (on 10q), and loss of p16 (on 9p) occur almost exclusively in glioblastoma and can provide means to distinguish anaplastic astrocytoma from glioblastoma.[51]
Acute lymphoblastic leukemia
Cytogenetics, the study of characteristic large changes in the chromosomes of cancer cells, has been increasingly recognized as an important predictor of outcome in acute lymphoblastic leukemia (ALL).[52]
NB: Balanced translocations cannot be detected by array-based karyotyping (see Limitations below).
Some cytogenetic subtypes have a worse prognosis than others. These include:
- A translocation between chromosomes 9 and 22, known as the Philadelphia chromosome, occurs in about 20% of adult and 5% in pediatric cases of ALL.
- A translocation between chromosomes 4 and 11 occurs in about 4% of cases and is most common in infants under 12 months.
- Not all translocations of chromosomes carry a poorer prognosis. Some translocations are relatively favorable. For example, Hyperdiploidy (>50 chromosomes) is a good prognostic factor.
- Genome-wide assessment of copy number changes can be done by conventional cytogenetics or virtual karyotyping. SNP array virtual karyotyping can detect copy number changes and LOH status, while arrayCGH can detect only copy number changes. Copy neutral LOH (acquired uniparental disomy) has been reported at key loci in ALL, such as CDKN2A gene at 9p, which have prognostic significance.[53][54][55] SNP array virtual karyotyping can readily detect copy neutral LOH. Array CGH, FISH, and conventional cytogenetics cannot detect copy neutral LOH.
| Cytogenetic change | Risk category |
|---|---|
| Philadelphia chromosome | Poor prognosis |
| t(4;11)(q21;q23) | Poor prognosis |
| t(8;14)(q24.1;q32) | Poor prognosis |
| Complex karyotype (more than four abnormalities) | Poor prognosis |
| Low hypodiploidy or near triploidy | Poor prognosis |
| High hyperdiploidy | Good prognosis |
| del(9p) | Good prognosis |
Correlation of prognosis with bone marrow cytogenetic finding in acute lymphoblastic leukemia
| Prognosis | Cytogenetic findings |
|---|---|
| Favorable | Hyperdiploidy > 50 ; t (12;21) |
| Intermediate | Hyperdioloidy 47 -50; Normal(diploidy); del (6q); Rearrangements of 8q24 |
| Unfavorable | Hypodiploidy-near haploidy; Near tetraploidy; del (17p); t (9;22); t (11q23) |
Unclassified ALL is considered to have an intermediate prognosis.[56]
Myelodysplastic syndrome
Myelodysplastic syndrome (MDS) has remarkable clinical, morphological, and genetic heterogeneity. Cytogenetics play a decisive role in the World Health Organization's classification-based International Prognostic Scoring System (IPSS) for MDS.[57][58]
- Good Prognosis: normal karyotype, isolated del(5q), isolated del(20q), -Y
- Poor Prognosis: complex abnormalities (i.e., >=3 abnormalities), −7 or del(7q)
- Intermediate Prognosis: all other abnormalities, including trisomy 8 and del(11q)
In a comparison of metaphase cytogenetics, FISH panel, and SNP array karyotyping for MDS, it was found that each technique provided a similar diagnostic yield. No single method detected all defects, and detection rates improved by ~5% when all three methods were used.[59]
Acquired UPD, which is not detectable by FISH or cytogenetics, has been reported at several key loci in MDS using SNP array karyotyping, including deletion of 7/7q.[60][61]
Myeloproliferative neoplasms/myeloproliferative disorders
Philadelphia chromosome–negative myeloproliferative neoplasms (MPNs) including polycythemia vera,essential thrombocythemia, and primary myelofibrosis show an inherent tendency for transformation into leukemia (MPN-blast phase),which is accompanied by acquisition of additional genomic lesions. In a study of 159 cases,[62] SNP-array analysis was able to capture practically all cytogenetic abnormalities and to uncover additional lesions with potentially important clinical implications.
- The number of genomic alterations was more than 2 to 3 times greater in the blast phase as in the chronic phase of the disease.
- Deletion of 17p (TP53)was significantly associated with prior exposure to hydroxyurea as well as a complex karyotype in samples with MPN-blast crisis. Interestingly, not only deletion, but also 17p copy neutral LOH, was associated with a complex karyotype, a poor prognostic marker in myeloid malignancies. Copy neutral LOH (acquired UPD)is readily detectably by SNP array karyotype, but not by cytogenetics, FISH, or arrayCGH.
- Blast phase patients with loss of chromosomal material on 7q showed poor survival. Loss of 7q is known to be predictive for rapid progression and poor response in AML therapy.MPN-blast phase patients with cytogenetically undetectable 7q copy neutral-LOH had comparable survival rates to those with 7/7q in their leukemic cells.
- 9p copy neutral-LOH with homozygous JAK2 mutation was also linked to an inferior outcome in MPN-blast crisis in comparison with patients with either heterozygous JAK2V617F or wild-type JAK2. In contrast to LOH on 17p, the prognostic impact of 9pCNN-LOH was independent of established risk factors such as 7/7q, 5q, or complex karyotype.
Colorectal cancer
Identification of biomarkers in colorectal cancer is particularly important for patients with stage II disease, where less than 20% have tumor recurrence. 18q LOH is an established biomarker associated with high risk of tumor recurrence in stage II colon cancer.[63] Figure 7 shows a SNP array karyotype of a colorectal carcinoma (whole genome view).
Colorectal cancers are classified into specific tumor phenotypes based on molecular profiles[63] which can be integrated with the results of other ancillary tests, such as microsatellite instability testing, IHC, and KRAS mutation status:
- Chromosomal instability (CIN) which have allelic imbalance at a number of chromosomal loci, including 5q, 8p, 17p, and 18q (Fig 7).
- Microsatellite instability (MSI) which tend to have diploid karyotypes.
Malignant rhabdoid tumors
Malignant rhabdoid tumors are rare, highly aggressive neoplasms found most commonly in infants and young children. Due to their heterogenous histologic features, diagnosis can often be difficult and misclassifications can occur. In these tumors, the INI1 gene (SMARCB1)on chromosome 22q functions as a classic tumor suppressor gene. Inactivation of INI1 can occur via deletion, mutation, or acquired UPD.[64]
In a recent study,[64] SNP array karyotyping identified deletions or LOH of 22q in 49/51 rhabdoid tumors. Of these, 14 were copy neutral LOH (or acquired UPD), which is detectable by SNP array karyotyping, but not by FISH, cytogenetics, or arrayCGH. MLPA detected a single exon homozygous deletion in one sample that was below the resolution of the SNP array.
SNP array karyotyping can be used to distinguish, for example, a medulloblastoma with an isochromosome 17q from a primary rhabdoid tumor with loss of 22q11.2. When indicated, molecular analysis of INI1 using MLPA and direct sequencing may then be employed. Once the tumor-associated changes are found, an analysis of germline DNA from the patient and the parents can be done to rule out an inherited or de novo germline mutation or deletion of INI1, so that appropriate recurrence risk assessments can be made.[64]
Uveal melanoma
The most important genetic alteration associated with poor prognosis in uveal melanoma is loss of an entire copy of Chromosome 3 (Monosomy 3), which is strongly correlated with metastatic spread.[65] Gains on chromosomes 6 and 8 are often used to refine the predictive value of the Monosomy 3 screen, with gain of 6p indicating a better prognosis and gain of 8q indicating a worse prognosis in disomy 3 tumors.[66] In rare instances, monosomy 3 tumors may duplicate the remaining copy of the chromosome to return to a disomic state referred to as isodisomy.[67] Isodisomy 3 is prognostically equivalent to monosomy 3, and both can be detected by tests for chromosome 3 loss of heterozygosity.[68]
Limitations
Unlike karyotypes obtained from conventional cytogenetics, virtual karyotypes are reconstructed by computer programs using signals obtained from disrupted DNA. In essence, the computer program will correct translocations when it lines up the signals in chromosomal order. Therefore, virtual karyotypes cannot detect balanced translocations and inversions. They also can only detect genetic aberrations in regions of the genome that are represented by probes on the array. In addition, virtual karyotypes generate a relative copy number normalized against a diploid genome, so tetraploid genomes will be condensed into a diploid space unless renormalization is performed. Renormalization requires an ancillary cell-based assay, such as FISH, if one is using arrayCGH. For karyotypes obtained from SNP-based arrays, tetraploidy can often be inferred from the maintenance of heterozygosity within a region of apparent copy number loss.[22] Low-level mosaicism or small subclones may not be detected by virtual karyotypes because the presence of normal cells in the sample will dampen the signal from the abnormal clone. The exact point of failure, in terms of the minimal percentage of neoplastic cells, will depend on the particular platform and algorithms used. Many copy number analysis software programs used to generate array-based karyotypes will falter with less than 25–30% tumor/abnormal cells in the sample. However, in oncology applications this limitation can be minimized by tumor enrichment strategies and software optimized for use with oncology samples. The analysis algorithms are evolving rapidly, and some are even designed to thrive on ‘normal clone contamination’,[69] so it is anticipated that this limitation will continue to dissipate.
See also
- DECIPHER, a Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources
References
- ↑ Digital karyotyping - Wang et al., 10.1073/pnas.202610899 - Proceedings of the National Academy of Sciences
- ↑ Shinawi M, Cheung SW (2008). "The array CGH and its clinical applications". Drug Discov Today. 13 (17–18): 760–70. doi:10.1016/j.drudis.2008.06.007. PMID 18617013.
- ↑ White M.J.D. 1973. The chromosomes. 6th ed, Chapman & Hall, London. p28
- ↑ Stebbins G.L. 1950. Variation and evolution in plants. Chapter XII: The Karyotype. Columbia University Press N.Y.
- ↑ Shaffer LG, Bejjani B (2006). "Medical applications of array CGH and the transformation of clinical cytogenetics". Cytogenet. Genome Res. 115 (3–4): 303–9. doi:10.1159/000095928. PMID 17124414.
- ↑ Edelmann L, Hirschhorn K (January 2009). "Clinical utility of array CGH for the detection of chromosomal imbalances associated with mental retardation and multiple congenital anomalies". Annals of the New York Academy of Sciences. 1151 (1): 157–66. doi:10.1111/j.1749-6632.2008.03610.x. PMID 19154522.
- ↑ Dutt A, Beroukhim R (January 2007). "Single nucleotide polymorphism array analysis of cancer". Current Opinion in Oncology. 19 (1): 43–9. doi:10.1097/CCO.0b013e328011a8c1. PMID 17133111.
- 1 2 3 Hagenkord JM, Parwani AV, Lyons-Weiler MA, Alvarez K, Amato R, Gatalica Z, Gonzalez-Berjon JM, Peterson L, Dhir R, Monzon FA (November 2008). "Virtual karyotyping with SNP microarrays reduces uncertainty in the diagnosis of renal epithelial tumors". Diagn Pathol. 3 (1): 44. doi:10.1186/1746-1596-3-44. PMC 2588560
 . PMID 18990225.
. PMID 18990225. - ↑ Beaudet AL, Belmont J (2008). "Array-based DNA diagnostics: let the revolution begin". Annu Rev Med. 59 (1): 113–29. doi:10.1146/annurev.med.59.012907.101800. PMID 17961075.
- 1 2 Monzon FA, Hagenkord JM, Lyons-Weiler MA, Balani JP, Parwani AV, Sciulli CM, Li J, Chandran UR, Bastacky SI, Dhir R. "Whole genome SNP arrays as a potential diagnostic tool for the detection of characteristic chromosomal aberrations in renal epithelial tumors". Mod Pathol. 21 (5): 599–608. doi:10.1038/modpathol.2008.20. PMID 18246049.
- ↑ Leary RJ, Lin JC, Cummins J, Boca S, Wood LD, Parsons DW, Jones S, Sjöblom T, Park BH, Parsons R, Willis J, Dawson D, Willson JK, Nikolskaya T, Nikolsky Y, Kopelovich L, Papadopoulos N, Pennacchio LA, Wang TL, Markowitz SD, Parmigiani G, Kinzler KW, Vogelstein B, Velculescu VE. "Integrated analysis of homozygous deletions, focal amplifications, and sequence alterations in breast and colorectal cancers". Proc Natl Acad Sci U S A. 105 (42): 16224–9. doi:10.1073/pnas.0808041105. PMC 2571022. PMID 18852474.
- 1 2 Lehmann S, Ogawa S, Raynaud SD, Sanada M, Nannya Y, Ticchioni M, Bastard C, Kawamata N, Koeffler HP. "Molecular allelokaryotyping of early-stage, untreated chronic lymphocytic leukemia". Cancer. 112 (6): 1296–305. doi:10.1002/cncr.23270. PMID 18246537.
- ↑ Vermeesch JR, Fiegler H, de Leeuw N, Szuhai K, Schoumans J, Ciccone R, Speleman F, Rauch A, Clayton-Smith J, Van Ravenswaaij C, Sanlaville D, Patsalis PC, Firth H, Devriendt K, Zuffardi O. "Guidelines for molecular karyotyping in constitutional genetic diagnosis". Eur J Hum Genet. 15 (11): 1105–14. doi:10.1038/sj.ejhg.5201896. PMID 17637806.
- ↑ Kulharya AS, Flannery DB, Norris K, Lovell C, Levy B, Velagaleti G (September 2008). "Fine mapping of breakpoints in two unrelated patients with rare overlapping interstitial deletions of 9q with mild dysmorphic features". American Journal of Medical Genetics. 146A (17): 2234–41. doi:10.1002/ajmg.a.32397. PMID 18666229.
- ↑ Nowakowska B; Stankiewicz P; Obersztyn E; Ou Z; Li J; Chinault AC; Smyk M; Borg K; Mazurczak T; Cheung SW; Bocian E. "Application of metaphase HR-CGH and targeted Chromosomal Microarray Analyses to genomic characterization of 116 patients with mental retardation and dysmorphic features". American Journal of Medical Genetics. 146A (18): 2361–9. doi:10.1002/ajmg.a.32475. PMID 18698622.
- ↑ Probst FJ, Roeder ER, Enciso VB, Ou Z, Cooper ML, Eng P, Li J, Gu Y, Stratton RF, Chinault AC, Shaw CA, Sutton VR, Cheung SW, Nelson DL. "Chromosomal microarray analysis (CMA) detects a large X chromosome deletion including FMR1, FMR2, and IDS in a female patient with mental retardation". American Journal of Medical Genetics. 143A (12): 1358–65. doi:10.1002/ajmg.a.31781. PMID 17506108.
- ↑ Hildebrandt, F; et al. (January 2009). "A systematic approach to mapping recessive disease genes in individuals from outbred populations". PLoS Genet. 5 (1): e1000353. doi:10.1371/journal.pgen.1000353.
- ↑ McQuillan R, Leutenegger AL, Abdel-Rahman R, Franklin CS, Pericic M, Barac-Lauc L, Smolej-Narancic N, Janicijevic B, Polasek O, Tenesa A, Macleod AK, Farrington SM, Rudan P, Hayward C, Vitart V, Rudan I, Wild SH, Dunlop MG, Wright AF, Campbell H, Wilson JF. "Runs of homozygosity in European populations". Am J Hum Genet. 83 (3): 359–72. doi:10.1016/j.ajhg.2008.08.007. PMC 2556426. PMID 18760389.
- ↑ Gondek LP, Tiu R, O'Keefe CL, Sekeres MA, Theil KS, Maciejewski J (February 2008). "Chromosomal lesions and uniparental disomy detected by SNP arrays in MDS, MDS/MPD, and MDS-derived AML". Blood. 111 (3): 1534–42. doi:10.1182/blood-2007-05-092304. PMC 2214746. PMID 17954704.
- ↑ Beroukhim R, Lin M, Park Y, Hao K, Zhao X, Garraway LA, Fox EA, Hochberg EP, Mellinghoff IK, Hofer MD, Descazeaud A, Rubin MA, Meyerson M, Wong WH, Sellers WR, Li C (May 2006). "Inferring loss-of-heterozygosity from unpaired tumors using high-density oligonucleotide SNP arrays". PLoS Comput. Biol. 2 (5): e41. doi:10.1371/journal.pcbi.0020041. PMC 1458964. PMID 16699594.
- ↑ Ishikawa S, Komura D, Tsuji S, Nishimura K, Yamamoto S, Panda B, Huang J, Fukayama M, Jones KW, Aburatani H. "Allelic dosage analysis with genotyping microarrays". Biochem Biophys Res Commun. 333 (4): 1309–14. doi:10.1016/j.bbrc.2005.06.040. PMID 15982637.
- 1 2 Lo KC, Bailey D, Burkhardt T, Gardina P, Turpaz Y, Cowell J (March 2008). "Comprehensive analysis of loss of heterozygosity events in glioblastoma using the 100K SNP mapping arrays and comparison with copy number abnormalities defined by BAC array comparative genomic hybridization". Genes Chromosomes Cancer. 47 (3): 221–37. doi:10.1002/gcc.20524. PMID 18050302.
- ↑ Mao X, Young BD, Lu Y (June 2007). "The application of single nucleotide polymorphism microarrays in cancer research". Curr Genomics. 8 (4): 219–28. doi:10.2174/138920207781386924. PMC 2430687. PMID 18645599.
- ↑ Janoueix-Lerosey I, Schleiermacher G, Michels E, et al. (March 2009). "Overall genomic pattern is a predictor of outcome in neuroblastoma". J. Clin. Oncol. 27 (7): 1026–33. doi:10.1200/JCO.2008.16.0630. PMID 19171713.
- ↑ Michels E, Vandesompele J, Hoebeeck J, Menten B, De Preter K, Laureys G, Van Roy N, Speleman F (2006). "Genome wide measurement of DNA copy number changes in neuroblastoma: dissecting amplicons and mapping losses, gains and breakpoints". Cytogenet. Genome Res. 115 (3–4): 273–282. doi:10.1159/000095924. PMID 17124410.
- ↑ Messahel B, Williams R, Ridolfi A, A'hern R, Warren W, Tinworth L, Hobson R, Al-Saadi R, Whyman G, Brundler MA, Kelsey A, Sebire N, Jones C, Vujanic G, Pritchard-Jones K, Children's Cancer and Leukaemia Group (CCLG) (March 2009). "Children's Cancer and Leukaemia Group (CCLG). Allele loss at 16q defines poorer prognosis Wilms tumour irrespective of treatment approach in the UKW1-3 clinical trials: a Children's Cancer and Leukaemia Group (CCLG) Study". Eur J Cancer. 45 (5): 819–26. doi:10.1016/j.ejca.2009.01.005. PMID 19231157.
- ↑ Grundy PE, Breslow NE, Li S, Perlman E, Beckwith JB, Ritchey ML, Shamberger RC, Haase GM, D'Angio GJ, Donaldson M, Coppes MJ, Malogolowkin M, Shearer P, Thomas PR, Macklis R, Tomlinson G, Huff V, Green DM, National Wilms Tumor Study Group (October 2005). "National Wilms Tumor Study Group. Loss of heterozygosity for chromosomes 1p and 16q is an adverse prognostic factor in favorable-histology Wilms tumor: a report from the National Wilms Tumor Study Group". J Clin Oncol. 23 (29): 7312–21. doi:10.1200/JCO.2005.01.2799. PMID 16129848.
- ↑ van den Berg, E; Störkel, S (2003). "Kidney: Clear cell renal cell carcinoma". Atlas Genet Cytogenet Oncol Haematol. 7 (3): 424–431. Retrieved 14 December 2010.
- ↑ Lyons-Weiler MA, Hagenkord JM, Sciulli CM, Dhir R, Monzon F (2008). "Optimization of the Affymetrix GeneChip Mapping 10K 2.0 Assay for Routine Clinical Use on Formalin Fixed Paraffin Embedded Tissues". Diag Mol Path. 17 (1): 3–13. doi:10.1097/PDM.0b013e31815aca30. PMID 18303412.
- ↑ Klatte T, Pantuck AJ, Said JW, Seligson DB, Rao NP, LaRochelle JC, Shuch B, Zisman A, Kabbinavar FF, Belldegrun AS (2009). "Cytogenetic and molecular tumor profiling for type 1 and type 2 papillary renal cell carcinoma.". Clinical Cancer Research. 15 (4): 1162–9. doi:10.1158/1078-0432.CCR-08-1229. PMID 19228721.
- ↑ Brunelli M, Eccher A, Gobbo S, Ficarra V, Novara G, Cossu-Rocca P, Bonetti F, Menestrina F, Cheng L, Eble JN, Martignoni G (2008). "Loss of chromosome 9p is an independent prognostic factor in patients with clear cell renal cell carcinoma.". Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 21 (1): 1–6. doi:10.1038/modpathol.3800967. PMID 17906617.
- ↑ Klatte T, Rao PN, de Martino M, LaRochelle J, Shuch B, Zomorodian N, Said J, Kabbinavar FF, Belldegrun AS, Pantuck AJ (2009). "Cytogenetic profile predicts prognosis of patients with clear cell renal cell carcinoma.". Journal of Clinical Oncology. 27 (5): 746–53. doi:10.1200/JCO.2007.15.8345. PMID 19124809.
- ↑ Szponar A, Zubakov D, Pawlak J, Jauch A, Kovacs G (2009). "Three genetic developmental stages of papillary renal cell tumors: duplication of chromosome 1q marks fatal progression.". International Journal of Cancer. Journal International Du Cancer. 124 (9): 2071–6. doi:10.1002/ijc.24180. PMID 19123481.
- ↑ Schwaenen C, Nessling M, Wessendorf S, Salvi T, Wrobel G, Radlwimmer B, Kestler HA, Haslinger C, Stilgenbauer S, Döhner H, Bentz M, Lichter P. "Automated array-based genomic profiling in chronic lymphocytic leukemia: development of a clinical tool and discovery of recurrent genomic alterations". Proc Natl Acad Sci U S A. 101 (4): 1039–44. doi:10.1073/pnas.0304717101. PMC 327147. PMID 14730057.
- ↑ Pfeifer D, Pantic M, Skatulla I, Rawluk J, Kreutz C, Martens UM, Fisch P, Timmer J, Veelken H. "Genome-wide analysis of DNA copy number changes and LOH in CLL using high-density SNP arrays". Blood. 109 (3): 1202–10. doi:10.1182/blood-2006-07-034256. PMID 17053054.
- ↑ Gunn SR; Mohammed MS; Gorre ME; Cotter PD; Kim J; Bahler DW; Preobrazhensky SN; Higgins RA; Bolla AR; Ismail SH; de Jong D; Eldering E; van Oers MH; Mellink CH; Keating MJ; Schlette EJ; Abruzzo LV; Robetorye RS (September 2008). "Whole-genome scanning by array comparative genomic hybridization as a clinical tool for risk assessment in chronic lymphocytic leukemia.". The Journal of molecular diagnostics : JMD. 10 (5): 442–451. doi:10.2353/jmoldx.2008.080033. PMC 2518739. PMID 18687794.
- ↑ Sargent R, Jones D, Abruzzo LV, Yao H, Bonderover J, Cisneros M, Wierda WG, Keating MJ, Luthra R (January 2009). "Customized oligonucleotide array-based comparative genomic hybridization as a clinical assay for genomic profiling of chronic lymphocytic leukemia". J Mol Diagn. 11 (1): 25–34. doi:10.2353/jmoldx.2009.080037. PMC 2607562. PMID 19074592.
- ↑ 2009 May;23(5):829-33
- ↑ Hagenkord JM, Monzon FA, Kash SF, Lilleberg S, Xie Q, Kant JA (2010). "Array-based karyotyping for prognostic assessment in chronic lymphocytic leukemia: performance comparison of affymetrix 10K2.0, 250K Nsp, and SNP6.0 arrays". J Mol Diagn. 12 (2): 184–96. doi:10.2353/jmoldx.2010.090118. PMC 2871725. PMID 20075210.
- ↑ Dohner H, Stilgenbauer S, Benner A, et al. (2000). "Genomic aberrations and survival in chronic lymphocytic leukemia.". NEJM. 343 (26): 1910–6. doi:10.1056/NEJM200012283432602. PMID 11136261.
- ↑ Hervé Avet-Loiseau; Cheng Li; Florence Magrangeas; Wilfried Gouraud; Catherine Charbonnel; Jean-Luc Harousseau; Michel Attal; Gerald Marit; Claire Mathiot; Thierry Facon; Philippe Moreau; Kenneth C. Anderson; Loïc Campion; Nikhil C. Munshi; Stéphane Minvielle (September 2009). "Prognostic significance of copy-number alterations in multiple myeloma.". Journal of Clinical Oncology. 27 (27): 4585–90. doi:10.1200/JCO.2008.20.6136. PMC 2754906. PMID 19687334.
- ↑ Pfister S; Remke M; Benner A; Mendrzyk F; Toedt G; Felsberg J; Wittmann A; Devens F; Gerber NU; Joos S; Kulozik A; Reifenberger G; Rutkowski S; Wiestler OD; Radlwimmer B; Scheurlen W; Lichter P; Korshunov A (April 2009). "Outcome Prediction in Pediatric Medulloblastoma based on DNA Copy Number Aberrations of Chromosomes 6q and 17q and the MYC and MYCN Loci". J Clin Oncol. 27 (10): 1627–1636. doi:10.1200/JCO.2008.17.9432. PMID 19255330.
- 1 2 Barbashina V, Salazar P, Holland EC, Rosenblum MK, Ladanyi M (1 February 2005). "Allelic losses at 1p36 and 19q13 in gliomas: correlation with histologic classification, definition of a 150-kb minimal deleted region on 1p36, and evaluation of CAMTA1 as a candidate tumor suppressor gene". Clin Cancer Res. 11 (3): 1119–28. PMID 15709179.
- ↑ Laigle-Donadey F, Benouaich-Amiel A, Hoang-Xuan K, Sanson M (2005). "[Molecular biology of oligodendroglial tumors]". Neuro-Chirurgie (in French). 51 (3–4 Pt 2): 260–8. doi:10.1016/s0028-3770(05)83487-3. PMID 16292170.
- ↑ Walker C, Haylock B, Husband D, et al. (2006). "Clinical use of genotype to predict chemosensitivity in oligodendroglial tumors". Neurology. 66 (11): 1661–7. doi:10.1212/01.wnl.0000218270.12495.9a. PMID 16769937.
- ↑ Jenkins RB, Blair H, Ballman KV, et al. (October 2006). "A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma". Cancer Res. 66 (20): 9852–61. doi:10.1158/0008-5472.CAN-06-1796. PMID 17047046.
- ↑ Ohgaki H, Eibl RH, Wiestler OD, Yasargil MG, Newcomb EW, Kleihues P (15 November 1991). "p53 mutations in nonastrocytic human brain tumors". Cancer Res. 51 (22): 6202–5. PMID 1933879.
- ↑ Ducray F, Idbaih A, de Reyniès A, et al. (2008). "Anaplastic oligodendrogliomas with 1p19q codeletion have a proneural gene expression profile". Mol. Cancer. 7 (1): 41. doi:10.1186/1476-4598-7-41. PMC 2415112. PMID 18492260.
- ↑ Dong Yin, Seishi Ogawa, Norihiko Kawamata, Patrizia Tunici, Gaetano Finocchiaro, Marica Eoli, Christian Ruckert, Thien Huynh, Gentao Liu, Motohiro Kato, Masashi Sanada, Anna Jauch, Martin Dugas, Keith L. Black, H. Phillip Koeffler (May 2009). "High-Resolution Genomic Copy Number Profiling of Glioblastoma Multiforme by Single Nucleotide Polymorphism DNA Microarray". Mol Cancer Res. 7 (5): 5. doi:10.1158/1541-7786.MCR-08-0270.
- ↑ Cancer Cytogenetics, 3rd Ed, Chapter 19, Tumors of the Nervous System, Wiley Blackwell 2009.
- ↑ Tumors of the Central Nervous System. Vol 7. Washington DC: American Registry of Pathology; 2007
- ↑ Moorman A, Harrison C, Buck G, Richards S, Secker-Walker L, Martineau M, Vance G, Cherry A, Higgins R, Fielding A, Foroni L, Paietta E, Tallman M, Litzow M, Wiernik P, Rowe J, Goldstone A, Dewald G (2007). "Karyotype is an independent prognostic factor in adult acute lymphoblastic leukemia (ALL): analysis of cytogenetic data from patients treated on the Medical Research Council (MRC) UKALLXII/Eastern Cooperative Oncology Group (ECOG) 2993 trial". Blood. 109 (8): 3189–97. doi:10.1182/blood-2006-10-051912. PMID 17170120.
- ↑ Kawamata N, Ogawa S, Zimmermann M, Kato M, Sanada M, Hemminki K, Yamatomo G, Nannya Y, Koehler R, Flohr T, Miller CW, Harbott J, Ludwig WD, Stanulla M, Schrappe M, Bartram CR, Koeffler HP (January 2008). "Molecular allelokaryotyping of pediatric acute lymphoblastic leukemias by high-resolution single nucleotide polymorphism oligonucleotide genomic microarray". Blood. 111 (2): 776–84. doi:10.1182/blood-2007-05-088310. PMC 2200831. PMID 17890455.
- ↑ Bungaro S, Dell'Orto MC, Zangrando A, Basso D, Gorletta T, Lo Nigro L, Leszl A, Young BD, Basso G, Bicciato S, Biondi A, te Kronnie G, Cazzaniga G (January 2009). "Integration of genomic and gene expression data of childhood ALL without known aberrations identifies subgroups with specific genetic hallmarks". Genes Chromosomes Cancer. 48 (1): 22–38. doi:10.1002/gcc.20616. PMID 18803328.
- ↑ Sulong S, Moorman AV, Irving JA, Strefford JC, Konn ZJ, Case MC, Minto L, Barber KE, Parker H, Wright SL, Stewart AR, Bailey S, Bown NP, Hall AG, Harrison CJ (January 2009). "A comprehensive analysis of the CDKN2A gene in childhood acute lymphoblastic leukemia reveals genomic deletion, copy number neutral loss of heterozygosity, and association with specific cytogenetic subgroups". Blood. 113 (1): 100–7. doi:10.1182/blood-2008-07-166801. PMID 18838613.
- ↑ Den Boer ML, van Slegtenhorst M, De Menezes RX, et al. (January 2009). "A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study". Lancet Oncol. 10 (2): 125–34. doi:10.1016/S1470-2045(08)70339-5. PMC 2707020. PMID 19138562.
- ↑ Hasse D (2008). "Cytogenetic features in myelodysplastic syndromes". Ann Hematol. 87 (7): 515–526. doi:10.1007/s00277-008-0483-y. PMC 2413090. PMID 18414863.
- ↑ WHO Classification of Tumours of Haematopoeitic and Lymphoid Tissues, Edited by Swerdlow SH, et al. IARC Press, 2008, Lyon.
- ↑ Makishima H, Rataul M, Gondek LP, Huh J, Cook JR, Theil KS, Sekeres MA, Kuczkowski E, O'Keefe C, Maciejewski JP (2010). "FISH and SNP-A karyotyping in myelodysplastic syndromes: Improving cytogenetic detection of del(5q), monosomy 7, del(7q), trisomy 8, and del(20q)". Leuk Res. 34 (4): 447–453. doi:10.1016/j.leukres.2009.08.023. PMC 2826525. PMID 19758696.
- ↑ Sanada, et al. "Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms." Nature 13 Aug 2009; 460, 904–909.
- ↑ Gondek LP, Tiu R, O'Keefe CL, Sekeres MA, Theil KS, MacIejewski JP (2008). "Chromosomal lesions and uniparental disomy detected by SNP arrays in MDS, MDS/MPD, and MDS-derived AML". Blood. 111 (3): 1534–42. doi:10.1182/blood-2007-05-092304. PMC 2214746. PMID 17954704.
- ↑ Thoennissen NH, Krug UO, Lee DH, Kawamata N, Iwanski GB, Lasho T, Weiss T, Nowak D, Koren-Michowitz M, Kato M, Sanada M, Shih LY, Nagler A, Raynaud SD, Müller-Tidow C, Mesa R, Haferlach T, Gilliland DG, Tefferi A, Ogawa S, Koeffler HP (April 2010). "Prevalence and prognostic impact of allelic imbalances associated with leukemic transformation of Philadelphia chromosome-negative myeloproliferative neoplasms". Blood. 14 (115): 2882–2890. doi:10.1182/blood-2009-07-235119. PMC 2854432. PMID 20068225.
- 1 2 Lenz HJ, "Established Biomarkers for Colorectal Carcinoma", American Society of Clinical Oncology Educational Book, 2009, p215-219.
- 1 2 3 Jackson EM, Sievert AJ, Gai X, Hakonarson H, Judkins AR, Tooke L, Perin JC, Xie H, Shaikh TH, Biegel JA. "Genomic analysis using high density single nucleotide polymorphism-based oligonucleotide arrays and multiplex ligation-dependent probe amplification provides comprehensive analysis of INI1/SMARCB1 in Malignant Rhabdoid Tumors". Clin Cancer Res. 15 (6): 1923–1930. doi:10.1158/1078-0432.CCR-08-2091. PMC 2668138. PMID 19276269.
- ↑ Prescher G, Bornfeld N, Hirche H, Horsthemke B, Jöckel KH, Becher R (1996). "Prognostic implications of monosomy 3 in uveal melanoma". Lancet. 347 (9010): 1222–1225. doi:10.1016/S0140-6736(96)90736-9. PMID 8622452.
- ↑ Damato BE, Dopierala J, Klaasen A, van Dijk M, Sibbring J, Coupland S (2009). "Multiplex Ligation-Dependent Probe Amplification of Uveal Melanoma: Correlation with Metastatic Death". Invest Ophthalmol Vis Sci. 50 (7): 3048–55. doi:10.1167/iovs.08-3165. PMID 19182252.
- ↑ White VA, McNeil BK, Horsman DE (1998). "Acquired homozygosity (isodisomy) of chromosome 3 in uveal melanoma". Cancer Genet Cytogenet. 102 (1): 40–45. doi:10.1016/S0165-4608(97)00290-2. PMID 9530338.
- ↑ Onken MD, Worley LA, Person E, Char DH, Bowcock AM, Harbour JW (2007). "Loss of heterozygosity of chromosome 3 detected with single nucleotide polymorphisms is superior to monosomy 3 for predicting metastasis in uveal melanoma". Clin Cancer Res. 13 (10): 2923–2937. doi:10.1158/1078-0432.CCR-06-2383. PMID 17504992.
- ↑ Yamamoto G, Nannya Y, Kato M, Sanada M, Levine RL, Kawamata N, Hangaishi A, Kurokawa M, Chiba S, Gilliland DG, Koeffler HP, Ogawa S (July 2007). "Highly sensitive method for genomewide detection of allelic composition in nonpaired, primary tumor specimens by use of affymetrix single-nucleotide-polymorphism genotyping microarrays". Am J Hum Genet. 81 (1): 114–26. doi:10.1086/518809. PMC 1950910. PMID 17564968.