Protein pKa calculations
In computational biology, protein pKa calculations are used to estimate the pKa values of amino acids as they exist within proteins. These calculations complement the pKa values reported for amino acids in their free state, and are used frequently within the fields of molecular modeling, structural bioinformatics, and computational biology.
Amino acid pKa values
pKa values of amino acid side chains play an important role in defining the pH-dependent characteristics of a protein. The pH-dependence of the activity displayed by enzymes and the pH-dependence of protein stability, for example, are properties that are determined by the pKa values of amino acid side chains.
The pKa values of an amino acid side chain in solution is typically inferred from the pKa values of model compounds (compounds that are similar to the side chains of amino acids). See Amino acid for the pKa values of all amino acid side chains inferred in such a way. There are also numerous experimental studies that have yielded such values, for example by use of NMR spectroscopy.
The table below lists the model pKa values that are often used in a protein pKa calculation, and contains a third column based on protein studies.[1]
| Amino Acid | pKa | pKa |
|---|---|---|
| Asp (D) | 3.9 | 4.0 |
| Glu (E) | 4.3 | 4.4 |
| Arg (R) | 12.0 | 13.5 |
| Lys (K) | 10.5 | 10.4 |
| His (H) | 6.08 | 6.8 |
| Cys (C) (–SH) | 8.28 | 8.3 |
| Tyr (Y) | 10.1 | 9.6 |
| N-term | 8.0 | |
| C-term | 3.6 |
The effect of the protein environment
When a protein folds, the titratable amino acids in the protein are transferred from a solution-like environment to an environment determined by the 3-dimensional structure of the protein. For example, in an unfolded protein an aspartic acid typically is in an environment which exposes the titratable side chain to water. When the protein folds the aspartic acid could find itself buried deep in the protein interior with no exposure to solvent.
Furthermore, in the folded protein the aspartic acid will be closer to other titratable groups in the protein and will also interact with permanent charges (e.g. ions) and dipoles in the protein. All of these effects alter the pKa value of the amino acid side chain, and pKa calculation methods generally calculate the effect of the protein environment on the model pKa value of an amino acid side chain.[2][3][4][5]
Typically the effects of the protein environment on the amino acid pKa value are divided into pH-independent effects and pH-dependent effects. The pH-independent effects (desolvation, interactions with permanent charges and dipoles) are added to the model pKa value to give the intrinsic pKa value. The pH-dependent effects cannot be added in the same straight-forward way and have to be accounted for using Boltzmann summation, Tanford–Roxby iterations or other methods.
The interplay of the intrinsic pKa values of a system with the electrostatic interaction energies between titratable groups can produce quite spectacular effects such as non-Henderson–Hasselbalch titration curves and even back-titration effects.[6]
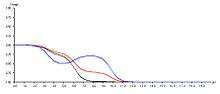
The image below shows a theoretical system consisting of three acidic residues. One group is displaying a back-titration event (blue group).
pKa calculation methods
Several software packages and webserver are available for the calculation of protein pKa values. See links below or this table
Using the Poisson–Boltzmann equation
Some methods are based on solutions to the Poisson–Boltzmann equation (PBE), often referred to as FDPB-based methods (FDPB is for "finite difference Poisson–Boltzmann"). The PBE is a modification of Poisson's equation that incorporates a description of the effect of solvent ions on the electrostatic field around a molecule.
The H++ web server, the pKD webserver, MCCE, Karlsberg+, PETIT and GMCT use the FDPB method to compute pKa values of amino acid side chains.
FDPB-based methods calculate the change in the pKa value of an amino acid side chain when that side chain is moved from a hypothetical fully solvated state to its position in the protein. To perform such a calculation, one needs theoretical methods that can calculate the effect of the protein interior on a pKa value, and knowledge of the pKa values of amino acid side chains in their fully solvated states.[2][3][4][5]
Empirical methods
A set of empirical rules relating the protein structure to the pKa values of ionizable residues have been developed by Li, Robertson, and Jensen. These rules form the basis for the web-accessible program called PROPKA for rapid predictions of pKa values. A recent empirical pKa prediction program was released by Tan KP et.al. with the online server DEPTH web server
Molecular dynamics (MD)-based methods
Molecular dynamics methods of calculating pKa values make it possible to include full flexibility of the titrated molecule.[7][8][9]
Molecular dynamics based methods are typically much more computationally expensive, and not necessarily more accurate, ways to predict pKa values than approaches based on the Poisson–Boltzmann equation. Limited conformational flexibility can also be realized within a continuum electrostatics approach, e.g., for considering multiple amino acid sidechain rotamers. In addition, current commonly used molecular force fields do not take electronic polarizability into account, which could be an important property in determining protonation energies.
Determining pKa values from titration curves or free energy calculations
From the titration of protonatable group, one can read the so-called pKa 1⁄2 which is equal to the pH value where the group is half-protonated. The pKa 1⁄2 is equal to the Henderson–Hasselbalch pKa (pKHH
a)
if the titration curve follows the Henderson–Hasselbalch equation.[10] Most pKa calculation methods silently assume that all titration curves are Henderson–Hasselbalch shaped, and pKa values in pKa calculation programs are therefore often determined in this way. In the general case of multiple interacting protonatable sites, the pKa 1⁄2 value is not thermodynamically meaningful. In contrast, the Henderson–Hasselbalch pKa value can be computed from the protonation free energy via

and is thus in turn related to the protonation free energy of the site via
.

The protonation free energy can in principle be computed from the protonation probability of the group ⟨x⟩(pH) which can be read from its titration curve
![{\displaystyle \Delta G^{\mathrm {prot} }(\mathrm {pH} )=-\mathrm {RT} \ln \left[{\frac {\langle x\rangle }{1-\langle x\rangle }}\right]}](../I/m/481d28b9f4ceef7840b1a2e9bce5c0e4b450067b.svg)
Titration curves can be computed within a continuum electrostatics approach with formally exact but more elaborate analytical or Monte Carlo (MC) methods, or inexact but fast approximate methods. MC methods that have been used to compute titration curves[11] are Metropolis MC[12] or Wang–Landau MC. Approximate methods that use a mean-field approach for computing titration curves are the Tanford–Roxby method and hybrids of this method that combine an exact statistical mechanics treatment within clusters of strongly interacting sites with a mean-field treatment of intercluster interactions.[13][14][15][16][17]
In practice, it can be difficult to obtain statistically converged and accurate protonation free energies from titration curves if ⟨x⟩ is close to a value of 1 or 0. In this case, one can use various free energy calculation methods to obtain the protonation free energy[11] such as biased Metropolis MC,[18] free-energy perturbation,[19][20] thermodynamic integration,[21][22][23] the non-equilibrium work method[24] or the Bennett acceptance ratio method.[25]
Note that the pKHH
a value does in general depend on the pH value.[26]
This dependence is small for weakly interacting groups like well solvated amino acid sidechains on the protein surface, but can be large for strongly interacting groups like those buried in enzyme active sites or integral membrane proteins.[27][28][29]
References
- ↑ Hass and Mulder (2015) Annu. Rev. Biophys. vol 44 pp. 53–75 doi 10.1146/annurev-biophys-083012-130351.
- 1 2 Bashford (2004) Front Biosci. vol. 9 pp. 1082–99 doi 10.2741/1187
- 1 2 Gunner et al. (2006) Biochim. Biophys. Acta vol. 1757 (8) pp. 942–68 doi 10.1016/j.bbabio.2006.06.005
- 1 2 Ullmann et al. (2008) Photosynth. Res. 97 vol. 112 pp. 33–55 doi 10.1007/s11120-008-9306-1
- 1 2 Antosiewicz et al. (2011) Mol. BioSyst. vol. 7 pp. 2923–2949 doi 10.1039/C1MB05170A
- ↑ A. Onufriev, D.A. Case and G. M. Ullmann (2001). Biochemistry 40: 3413–3419 doi 10.1021/bi002740q
- ↑ Donnini et al. (2011) J. Chem. Theory Comp. vol 7 pp. 1962–78 doi 10.1021/ct200061r.
- ↑ Wallace et al. (2011) J. Chem. Theory Comp. vol 7 pp. 2617–2629 doi 10.1021/ct200146j.
- ↑ Goh et al. (2012) J. Chem. Theory Comp. vol 8 pp. 36–46 doi 10.1021/ct2006314.
- ↑ Ullmann (2003) J. Phys. Chem. B vol 107 pp. 1263–71 doi 10.1021/jp026454v.
- 1 2 Ullmann et al. (2012) J. Comput. Chem. vol 33 pp. 887–900 doi 10.1002/jcc.22919
- ↑ Beroza et al. (1991) Proc. Natl. Acad. Sci. USA vol 88 pp. 5804–5808 doi 10.1073/pnas.88.13.5804
- ↑ Tanford and Roxby (1972) Biochemistry vol 11 pp. 2192–2198 doi 10.1021/bi00761a029
- ↑ Bashford and Karplus (1991) J. Phys. Chem. vol 95 pp. 9556–61 doi 10.1021/j100176a093
- ↑ Gilson (1993) Proteins vol 15 pp. 266–82 doi 10.1002/prot.340150305
- ↑ Antosiewicz et al. (1994) J. Mol. Biol. vol 238 pp. 415–36 doi 10.1006/jmbi.1994.1301
- ↑ Spassov and Bashford (1999) J. Comput. Chem. vol 20 pp. 1091–1111 doi 10.1002/(SICI)1096-987X(199908)20:11<1091::AID-JCC1>3.0.CO;2-3
- ↑ Beroza et al. (1995) Biophys. J. vol 68 pp. 2233–2250 doi 10.1016/S0006-3495(95)80406-6
- ↑ Zwanzig (1954) J. Chem. Phys. vol 22 pp. 1420–1426 doi 10.1063/1.1740409
- ↑ Ullmann et al. 2011 J. Phys. Chem. B. vol 68 pp. 507–521 doi 10.1021/jp1093838
- ↑ Kirkwood (1935) J. Chem. Phys. vol 2 pp. 300–313 doi 10.1063/1.1749657
- ↑ Bruckner and Boresch (2011) J. Comput. Chem. vol 32 pp. 1303–1319 doi 10.1002/jcc.21713
- ↑ Bruckner and Boresch (2011) J. Comput. Chem. vol 32 pp. 1320–1333 doi 10.1002/jcc.21712
- ↑ Jarzynski (1997) Phys. Rev. E vol pp. 2233–2250 doi 10.1103/PhysRevE.56.5018
- ↑ Bennett (1976) J. Comput. Phys. vol 22 pp. 245–268 doi 10.1016/0021-9991(76)90078-4
- ↑ Bombarda et al. (2010) J. Phys. Chem. B vol 114 pp. 1994–2003 doi 10.1021/jp908926w.
- ↑ Bashford and Gerwert (1992) J. Mol. Biol. vol 224 pp. 473–86 doi 10.1016/0022-2836(92)91009-E
- ↑ Spassov et al. (2001) J. Mol. Biol. vol 312 pp. 203–19 doi 10.1006/jmbi.2001.4902
- ↑ Ullmann et al. (2011) J. Phys. Chem. B vol 115 pp. 10346–59 doi 10.1021/jp204644h
Software for protein pKa calculations
- AccelrysPKA Accelrys CHARMm based pKa calculation
- H++ Poisson–Boltzmann based pKa calculations
- MCCE Multi-Conformation Continuum Electrostatics
- Karlsberg+ pKa computation with multiple pH adapted conformations
- PETIT Proton and Electron TITration
- GMCT Generalized Monte Carlo Titration
- pKD server pKa calculations and pKa value redesign
- PROPKA Empirical calculation of pKa values
- DEPTH web server Empirical calculation of pKa values using Residue Depth as a major feature
Analysis and teaching software
- pKaTool interactive graphical tool for analysing systems of titratable groups
- H++ webserver