Polystannane
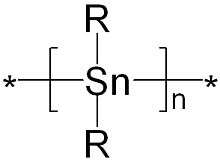
Polystannanes are organotin compounds with the formula (R2Sn)n. These polymers have been of intermittent academic interest; they are unusual because heavy elements comprise the backbone. Structurally related but better characterized (and more useful) are the polysilanes (R2Si)n.
History
Oligo- or polystannanes were first described by Löwig in 1852,[1] only 2 years after Edward Frankland's report on the isolation of the first organotin compounds.[2] Löwig' route involved treating an Sn/K and Sn/Na alloys with iodoethane, in the presence of quartz sand which was used to control the reaction rate. Products with elemental compositions close to those of oligo(diethylstannane)s or poly(diethylstannane) were obtained. Cahours obtained similar products and attributed the formation of the so-called "stannic ethyl" to a reaction of the Wurtz type.[3] Already in 1858, "stannic ethyl" was formulated as a polymeric compound denoted with the composition n(SnC4H5).[4] In 1917 Grüttner,[5] who reinvestigated results on hexaethyl-distannanes(H5C2)3Sn-Sn(C2H5)3 (reported by Ladenburg in 1870) confirmed the presence of Sn-Sn bonds and predicated for the first time that tin could form chain like compounds.[6] In 1943, it was postulated that “diphenyltin” exists as a type of polymeric material because of its yellow color [8], and indeed a bathochromic shift of the wavelength at maximum absorption with increasing number of Sn atoms was found later in the case of oligo(dibutylstannane)s comprising up to 15 Sn atoms.[7]
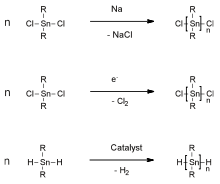
The Wurtz reaction is still used for the preparation of poly(dialkylstannane)s. Treatment of dialkyltin dichlorides with sodium lead to polystannanes of high molar mass, however, in low yields and with formation of (cyclic) oligomers. [11-16]. Other efforts to prepare high molar mass polystannanes by electrochemical reactions[8] or by catalytic dehydropolymerization of dialkylstannanes (R2SnH2) were also made [16].[9] Unfortunately, frequently, the polymers prepared by those methods were not isolated and typically contained significant fractions of cyclic oligomers.
Linear polystannanes
 Synthesis of pure linear poly(dibutylstannane).
Synthesis of pure linear poly(dibutylstannane). Opical micrographs (crossed polarizers) of an oriented film of poly(3-metylbutylstannane) produced by shearing the material at room temperature, top at 45° and bottom 90° in respect to the polarizers.
Opical micrographs (crossed polarizers) of an oriented film of poly(3-metylbutylstannane) produced by shearing the material at room temperature, top at 45° and bottom 90° in respect to the polarizers.
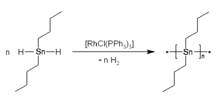
Dialkytin dihydrides (R2SnH2) were reported in 2005 to undergo dehydropolymerization in the presence of Wilkinson’s catalyst. This method afforded polystannanes without detectable amounts of "cyclic"-byproducts. The polymers were yellow with number average molar masses of 10 to 70 kg/mol and a polydispersity of 2 – 3.[10] By variation of the catalyst concentration the molar masses of the synthesized polymers could be adjusted. A strong influence of the temperature on the degree of conversion was observed. Determination of the molar mass at different degrees of conversion indicated that polymerization did not proceed according to a statistical condensation mechanism, but, likely, by growth onto the catalyst, e.g. by insertion of SnR2-like units.
The poly(dialkylstannane)s were found to be thermotropic and displayed first-order phase transitions from one liquid-crystalline phase into another or directly to the isotropic state, depending on the length of the side groups. More specifically, poly(dibutylstannane) for example showed an endothermic phase transition at ~0 °C from a rectangular to a pure nematic phase, as determined by X-ray diffraction.[11]
As expected, polystannanes were semi-conductive. Temperature-dependent, time-resolved pulse radiolysis microwave conductivity measurements of poly(dibutylstannane) yielded values of charge-carrier mobilities of 0.1 to 0.03 cm2 V−1 s−1, which are similar to those found for pi-bond-conjugated carbon-based polymers. By partial oxidation of the material with SbF5 conductivities of 0.3 S cm−1 could be monitored.[12]
The liquid-crystalline characteristics of the poly(dialkylstannane)s permitted facile orientation of these macromolecules, for instance, by mechanical shearing or tensile drawing of blends with poly(ethylene). Poly(dialkylstannane)s with short side groups invariably arranged parallel to the external orientation direction, while the polymers with longer side groups had a tendency to order themselves perpendicular to that axis.
References
- ↑ C. Löwig, Mitt. Naturforsch. Ges. Zürich, 1852, 2, 556.
- ↑ Edward Frankland|, Q. J. Chem. Soc., 1850, 2, 263.
- ↑ A. Cahours, Ann. Chem. Pharm. (Liebig’s Ann.), 1860, 114, 227A. Cahours, Ann. Chim. Phys., Sér. 3, 1860, 58, 5.
- ↑ A. Strecker, Ann. Chem. Pharm. (Liebig’s Ann.), 1858, 105, 306.
- ↑ G. Grüttner, Ber. Deutsch. Chem. Gesell., 1917, 50, 1808.
- ↑ A. Ladenburg, Ber. Deutsch. Chem. Gesell., 1870, 3, 353.
- ↑ L. R. Sita, K. W. Terry, K. Shibata, J. Am. Chem. Soc., 1995, 117, 8049.
- ↑ M. Okano, N. Matsumoto, M. Arakawa, T. Tsuruta, H. Hamano, Chem. Commun., 1998, 1799. M. Okano, K. Watanabe, Electrochem. Commun., 2000, 2, 471.
- ↑ T. Imori, T. D. Tilley, J. Chem. Soc., Chem. Commun., 1993, 1607; V. Y. Lu, T. D. Tilley, Macromolecules, 2000, 33, 2403; H. G. Woo, J. M. Park, S. J. Song, S. Y. Yang, I. S. Kim, W. G. Kim, Bull. Korean Chem. Soc., 1997, 18, 1291; H. G. Woo, S. J. Song, B. H. Kim, Bull. Korean Chem. Soc., 1998, 19, 1161.
- ↑ F. Choffat, P. Smith, W. Caseri, J. Mater. Chem., 2005, 15, 1789.
- ↑ F. Choffat, S. Käser, P. Wolfer, D. Schmid, R. Mezzenga, P. Smith, W. Caseri, Macromolecules, 2007, 40, 7878.
- ↑ T. Imori, V. Lu, H. Cai, T. D. Tilley, J. Am. Chem. Soc., 1995, 117, 9931.
External links
- Fabien Choffat (2007) Polystannane, Doctoral dissertation, Swiss Federal Institute of Technology, Zürich.