Paired-end tag
Paired-end tags (PET) (sometimes "Paired-End diTags", or simply "ditags") are the short sequences at the 5’ and 3’ ends of a DNA fragment which are unique enough that they (theoretically) exist together only once in a genome, therefore making the sequence of the DNA in between them available upon search (if full-genome sequence data is available) or upon further sequencing (since tag sites are unique enough to serve as primer annealing sites). Paired-end tags (PET) exist in PET libraries with the intervening DNA absent, that is, a PET "represents" a larger fragment of genomic or cDNA by consisting of a short 5' linker sequence, a short 5' sequence tag, a short 3' sequence tag, and a short 3' linker sequence. It was shown conceptually that 13 base pairs are sufficient to map tags uniquely.[1] However, longer sequences are more practical for mapping reads uniquely. The endonucleases (discussed below) used to produce PETs give longer tags (18/20 base pairs and 25/27 base pairs) but sequences of 50–100 base pairs would be optimal for both mapping and cost efficiency.[1] After extracting the PETs from many DNA fragments, they are linked (concatenated) together for efficient sequencing. On average, 20–30 tags could be sequenced with the Sanger method, which has a longer read length.[1] Since the tag sequences are short, individual PETs are well suited for next-generation sequencing that has short read lengths and higher throughput. The main advantages of PET sequencing are its reduced cost by sequencing only short fragments, detection of structural variants in the genome, and increased specificity when aligning back to the genome compared to single tags, which involves only one end of the DNA fragment.
Constructing the PET library
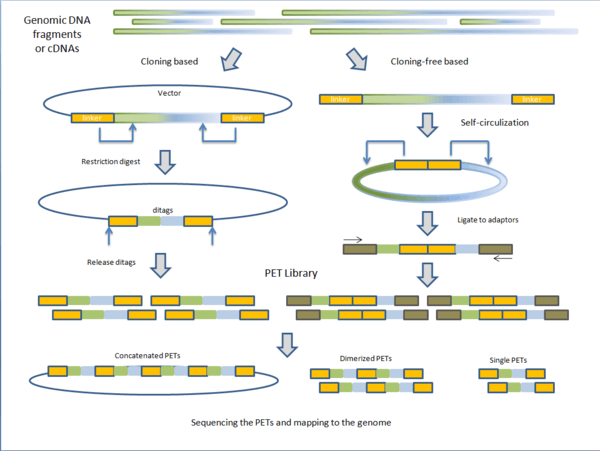
PET libraries are typically prepared in two general methods: cloning based and cloning-free based.
Cloning based
Fragmented genomic DNA or complementary DNA (cDNA) of interest is cloned into plasmid vectors. The cloning sites are flanked with adaptor sequences that contain restriction sites for endonucleases (discussed below). Inserts are ligated to the plasmid vectors and individual vectors are then transformed into E. coli making the PET library. PET sequences are obtained by purifying plasmid and digesting with specific endonuclease leaving two short sequences on the ends of the vectors. Under intramolecular (dilute) conditions, vectors are re-circularized and ligated, leaving only the ditags in the vector. The sequences unique to the clone are now paired together. Depending on the next-generation sequencing technique, PET sequences can be left singular, dimerized, or concatenated into long chains.[1]
Cloning-free based
Instead of cloning, adaptors containing the endonuclease sequence are ligated to the ends of fragmented genomic DNA or cDNA. The molecules are then self-circularized and digested with endonuclease, releasing the PET.[1] Before sequencing, these PETs are ligated to adaptors to which PCR primers anneal for amplification. The advantage of cloning based construction of the library is that it maintains the fragments or cDNA intact for future use. However, the construction process is much longer than the cloning-free method. Variations on library construction have been produced by next-generation sequencing companies to suit their respective technologies.[1]
Endonucleases
Unlike other endonucleases, the MmeI (type IIS) and EcoP15I (type III) restriction endonucleases cut downstream of their target binding sites. MmeI cuts 18/20 base pairs downstream [2] and EcoP15I cuts 25/27 base pairs downstream.[3] As these restriction enzymes bind at their target sequences located in the adaptors, they cut and release vectors that contain short sequences of the fragment or cDNA ligated to them, producing PETs.
PET applications
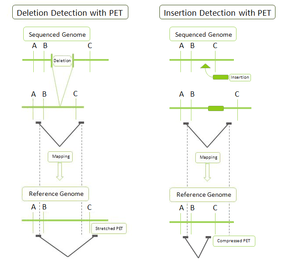
- DNA-PET: Because PET represent connectivity between the tags, the use of PET in genome re-sequencing has advantages over the use of single reads. This application is called pairwise end sequencing, known colloquially as double-barrel shotgun sequencing. Anchoring one half of the pair uniquely to a single location in the genome allows mapping of the other half that is ambiguous. Ambiguous reads are those that map to more than a single location. This increased efficiency reduces the cost of sequencing as these ambiguous sequences, or reads, would normally be discarded. The connectivity of PET sequences also allows detection of structural variations: insertions, deletions, duplications, inversions, translocations.[1][4] During the construction of the PET library, the fragments can be selected to all be of a certain size. After mapping, the PET sequences are thus expected to be consistently a particular distance away from each other. A discrepancy from this distance indicates a structural variation between the PET sequences. For example (Figure on the right): a deletion in the sequenced genome will have reads that map further away than expected in the reference genome as the reference genome will have a segment of DNA that is not present in the sequenced genome.
- ChIP-PET: The combined use of chromatin immunoprecipitation (ChIP) and PET is used to detect regions of DNA bound by a protein of interest. ChIP-PET has the advantage over single read sequencing by reducing ambiguity of the reads generated. The advantage over chip hybridization (ChIP-Chip) is that hybridization tiling arrays do not have the statistical sensitivity that sequence reads have. However, ChIP-PET, ChIP-Seq[5][6][7] and ChIP-chip[8] have all been highly successful.[1]
- ChIA-PET: The application of PET sequencing on chromatin interaction analysis. It is a genome-wide strategy for finding de novo long-range interactions between DNA elements bound by protein factors.[9] The first ChIA-PET was developed by Fullwood et al.. (2009)[9] to generate a map of the interactions between chromatin bound by oestrogen receptor α (ER-α) in oestrogen-treated human breast adenocarcinoma cells.[9] ChIA-PET is an unbiased way to analyze interactions and higher-order chromatin structures because it can detect interactions between unknown DNA elements. In contrast, 3C and 4C methods are used to detect interactions involving a specific target region in the genome. ChIA-PET is similar to finding fusion genes through RNA-PET in that the paired tags map to different regions in the genome.[1] However, ChIA-PET involves artificial ligations between different DNA fragments located at different genomic regions, rather than naturally occurring fusion between two genomic regions as in RNA-PET.
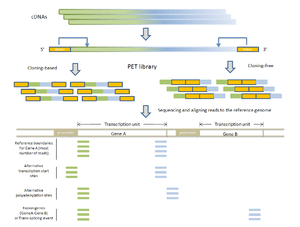
- RNA-PET: This application is used for studying the transcriptome: transcripts, gene structures, and gene expressions.[1][10] The PET library is generated using full length cDNAs, so the ditags represent the 5’ capped and the 3’ polyA tail signatures of individual transcripts. Therefore, RNA-PET is especially useful for demarcating the boundaries of transcription units. This will help identify alternative transcription start sites and polyadenylation sites of genes.[1] RNA-PET could also be used to detect fusion genes and trans-splicing, but further experiment is needed to distinguish between them.[11] Other methods of finding the boundaries of transcripts include the single-tag strategies CAGE, SAGE, and the most recent SuperSAGE, with the CAGE and 5’ SAGE defining the transcription start sites and the 3’ SAGE defining the polyadenylation sites.[1] The advantages of PET sequencing over these methods are that PET identify both ends of the transcripts and, at the same time, provide more specificity when mapping back to the genome. Sequencing the cDNAs can reveal the structures of transcripts in great details, but this approach is much more expensive than RNA-PET sequencing, especially for characterizing the whole transcriptome.[10] The major limitation of RNA-PET is the lack of information regarding the organization of the internal exons of transcripts. Therefore, RNA-PET is not suitable for detecting alternative splicing. In addition, if the cloning procedure is used construct the cDNA library before generating the PETs, cDNAs that are difficult to clone (for example, because of long transcripts) would have lower coverage.[10] Similarly, transcripts (or transcript isoforms) with low expression level would likely be under-represented as well.
References
- 1 2 3 4 5 6 7 8 9 10 11 12 Fullwood MJ, Wei CL, Liu ET, Ruan Y. 2009. Next-Generation DNA sequencing of paired-end tags (PET) for transcriptome and genome analyses. Genome Research. 19:521–532. PMID 19339662
- ↑ Morgan RD, Bhatia TK, Lovasco L, Davis TB. 2008. MmeI: A minimal Type II restriction-modification system that only modifies one DNA strand for host protection. Nucleic Acids Res. 36:6558–6570.
- ↑ Matsumura H, Reich S, Ito A, Saitoh H, Kamoun S, Winter P, Kahl G, Reuter M, Kruger DH, Terauchi R. 2003. Gene expression analysis of plant host-pathogen interactions by SuperSAGE. Proc. Natl. Acad. Sci. 100:15718–15723.
- ↑ McKernan et al. 2009. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Research. 19:1527-1541.
- ↑ Barski A, Cuddapa S, Cui K, Roh TY, Schones DE,Wang Z,Wei G, Chepelev I, Zhao K. 2007. High-resolution profiling of histone methylations in the human genome. Cell. 129:823–837.
- ↑ Johnson DS, Mortazavi A, Myers RM, Wold B. 2007. Genomewide mapping of in vivo protein-DNA interactions. Science. 316:1497–1502.
- ↑ Chen X et al. 2008. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133: 1106–1117.
- ↑ Wu J, Smith LT, Plass C, Huang TH. 2006. ChIP-chip comes of age for genome-wide functional analysis. Cancer Res. 66(14): 6899–902
- 1 2 3 Fullwood MJ, Liu MH, Pan YF et al. 2009. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature. 462: 58-64.
- 1 2 3 Ng P, Wei CL, Sung WK et al. 2005. Gene identification signature (GIS) analysis for transcriptome characterization and genome annotation. Nat. Methods. 2: 105–111.
- ↑ Ruan Y, Ooi HS, Choo SW et al. 2007. Fusion transcripts and transcribed retrotransposed loci discovered through comprehensive transcriptome analysis using Paired-End diTags (PETs). Genome Res. 17: 828–838.