Mucolipidosis type IV
| Mucolipidosis type IV | |
|---|---|
| Classification and external resources | |
| Specialty | endocrinology |
| ICD-10 | E75.1 |
| OMIM | 252650 |
| DiseasesDB | 32693 |
| MeSH | D009081 |
| GeneReviews | |
Mucolipidosis type IV (ML IV or ML4) is an autosomal recessive lysosomal storage disorder. Individuals with the disorder have many symptoms including delayed psychomotor development and various ocular aberrations. The disorder is caused by mutations in the MCOLN1 gene, which encodes a non-selective cation channel, mucolipin1.[1][2] These mutations disrupt cellular functions and lead to a neurodevelopmental disorder through an unknown mechanism. Researchers dispute the physiological role of the protein product and which ion it transports.[3]
Symptoms and Signs
Most patients with ML IV show psychomotor retardation (i.e., delayed development of movement and coordination), corneal opacity, retinal degeneration and other ophthalmological abnormalities. Other symptoms include agenesis of the corpus callosum, iron deficiency resulting from an absence of acid secretion in the stomach, achlorhydria. Achlorhydria in these patients results in an increase in blood gastrin levels. These symptoms typically manifest early in life (within the first year). After disease onset there occurs a period of stability, typically lasting two to three decades during which very little disease progression occurs.[4]
Pathophysiology
Mucolipin1 is thought to be localized in endosomes. An important property of mucolipin1 is that decreasing pH (acidification) results in deactivation of the protein, likely through an assembly defect. There are at least 29 known mutations in MCOLN1, located throughout the gene.[5] Many of the known mutations result in no expression of mucolipin1. Milder mutations, such as ΔF408 and V446L, produce a dysfunctioning form of the cation channel.[6] Mutations that alter only the C-terminal of the protein also result in a mild phenotype of the disorder, usually sparing the brain.[7] ML IV causes affected cells to accumulate auto-fluorescent vacuoles considered to be aberrant lysosomes.[8] Several evidences exist for a defect in both exocytosis and endocytosis.[9] There are conflicting indications of abnormal lysosomal pH in MLIV. It is not yet clear why these abnormalities will cause incomplete development of the brain, achlorhydria, and failure in the maintenance of retinal tissue.
Treatment/Management
There is no specific treatment to this disorder. However, several symptoms may be alleviated. For instance, anemia is treated by iron supplements. Some of the movement deficiencies may be corrected with orthopedic intervention. The corneal clouding can be, at least, temporarily corrected by corneal transplantation. See the equivalent section in the main mucolipidosis article.
Epidemiology
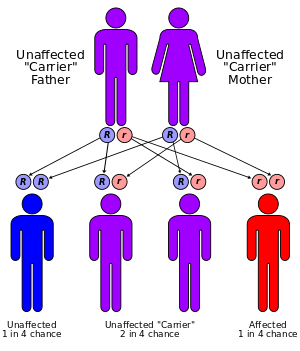
Mucolipidosis type IV is severely under-diagnosed. It is often misdiagnosed as cerebral palsy. In the Ashkenazi Jewish population there are two severe mutations with a higher carrier frequency[10] of 1:90 to 1:100.[11]
References
- ↑ Nilius, B.; Owsianik, G.; Voets, T.; Peters, J. A. (2007). "Transient Receptor Potential Cation Channels in Disease". Physiological Reviews. 87 (1): 165–217. doi:10.1152/physrev.00021.2006. PMID 17237345.
- ↑ Sun, M.; Goldin, E; Stahl, S; Falardeau, J. L.; Kennedy, J. C.; Acierno Jr, J. S.; Bove, C; Kaneski, C. R.; Nagle, J; Bromley, M. C.; Colman, M; Schiffmann, R; Slaugenhaupt, S. A. (2000). "Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel". Human Molecular Genetics. 9 (17): 2471–8. doi:10.1093/hmg/9.17.2471. PMID 11030752.
- ↑ Dong, Xian-Ping; Cheng, Xiping; Mills, Eric; Delling, Markus; Wang, Fudi; Kurz, Tino; Xu, Haoxing (2008). "The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel". Nature. 455 (7215): 992–6. Bibcode:2008Natur.455..992D. doi:10.1038/nature07311. PMID 18794901.
- ↑ Wakabayashi, K.; Gustafson, A. M.; Sidransky, E.; Goldin, E. (2011). "Mucolipidosis type IV: An update". Molecular Genetics and Metabolism. 104 (3): 206–213. doi:10.1016/j.ymgme.2011.06.006. PMID 21763169.
- ↑ Goldin E, Slaugenhaupt SA, Smith J, Schiffmann R (2005) Mucolipidosis IV, GeneReviews at GeneTests: Medical Genetics Information Resource http://www.ncbi.nlm.nih.gov/books/NBK1214/
- ↑ Altarescu, G.; Sun, M.; Moore, D. F.; Smith, J. A.; Wiggs, E. A.; Solomon, B. I.; Patronas, N. J.; Frei, K. P.; Gupta, S.; Kaneski, C. R.; Quarrell, O. W.; Slaugenhaupt, S. A.; Goldin, E.; Schiffmann, R. (2002). "The neurogenetics of mucolipidosis type IV". Neurology. 59 (3): 306–13. doi:10.1212/wnl.59.3.306. PMID 12182165.
- ↑ Goldin, E.; Caruso, R. C.; Benko, W.; Kaneski, C. R.; Stahl, S.; Schiffmann, R. (2008). "Isolated Ocular Disease is Associated with Decreased Mucolipin-1 Channel Conductance". Investigative Ophthalmology & Visual Science. 49 (7): 3134–42. doi:10.1167/iovs.07-1649. PMID 18326692.
- ↑ Goldin, Ehud; Blanchette-Mackie, E Joan; Dwyer, Nancy K; Pentchev, Peter G; Brady, Roscoe O (1995). "Cultured Skin Fibroblasts Derived from Patients with Mucolipidosis 4 Are Auto-Fluorescent". Pediatric Research. 37 (6): 687–92. doi:10.1203/00006450-199506000-00003. PMID 7651750.
- ↑ Chen, C. S.; Bach, G; Pagano, R. E. (1998). "Abnormal transport along the lysosomal pathway in Mucolipidosis, type IV disease". Proceedings of the National Academy of Sciences of the United States of America. 95 (11): 6373–6378. Bibcode:1998PNAS...95.6373C. doi:10.1073/pnas.95.11.6373. PMC 27719
 . PMID 9600972.
. PMID 9600972. - ↑ Bach, Gideon; Webb, Michael B.T.; Bargal, Ruth; Zeigler, Marcia; Ekstein, Joseph (2005). "The frequency of mucolipidosis type IV in the Ashkenazi Jewish population and the identification of 3 novel MCOLN1 mutations". Human Mutation. 26 (6): 591. doi:10.1002/humu.9385. PMID 16287144.
- ↑ Bargal, Ruth; Avidan, Nili; Olender, Tzvia; Ben Asher, Edna; Zeigler, Marcia; Raas-Rothschild, Annick; Frumkin, Ayala; Ben-Yoseph, Omer; Friedlender, Yechiel; Lancet, Doron; Bach, Gideon (2001). "Mucolipidosis type IV: NovelMCOLN1 mutations in Jewish and non-Jewish patients and the frequency of the disease in the Ashkenazi Jewish population". Human Mutation. 17 (5): 397–402. doi:10.1002/humu.1115. PMID 11317355.
External links
- Mucolipidosis type 4 at NIH's Office of Rare Diseases
- Mucolipidosis IV Foundation
- Gene Test Diagnostic Organization