Lafora disease
| Lafora disease | |
|---|---|
.jpg) | |
| Gonzalo Rodríguez Lafora, discoverer of the disease | |
| Classification and external resources | |
| Specialty | neurology |
| ICD-10 | G40.3 |
| ICD-9-CM | 333.2 |
| OMIM | 254780 |
| DiseasesDB | 30834 |
| MeSH | D020192 |
Lafora disease, also called Lafora progressive myoclonic epilepsy or MELF,[1] is a fatal autosomal recessive[2] genetic disorder characterized by the presence of inclusion bodies, known as Lafora bodies, within the cytoplasm of the cells of the heart, liver, muscle, and skin.[3]:545 In a later study, Lafora disease has been and is now viewed as a neurodegenerative disease, since prior to the actual formation of Lafora bodies there has been seen to be an impairment in the development of cerebral cortical neurons. It was further concluded that Lafora disease is a complex neurodegenerative disease and also a glycogen metabolism disorder.[4]
Most patients with this disease do not live past the age of twenty-five, and death within ten years of symptoms is usually inevitable.[5] At present, there is no cure or treatment for this disease.
Signs and symptoms
Patients develop the first symptoms mainly during adolescence. Major problems include seizures, drop attacks, myoclonus, ataxia, and, most significantly, a quickly-developing and severe dementia.[2]
Genetics
Lafora disease is an autosomal recessive disorder, caused by mutations in one of three known genes: EPM2A, EPM2B, and NHLRC1.[6][7][8]( edit : two genes: the EPM2A gene and the NHLRC1 gene which was previously known as the EPM2B gene. A third gene - PRDM8 gene- MAY cause an early onset form of the disease ). EPM2A codes for the protein laforin, a dual-specificity phosphatase with a carbohydrate binding domain (CBM-20). Vertebrates have only one such protein with DSP domain as well as CBM-20 domain. EPM2B encodes the protein malin, an E3 ubiquitin ligase. All three discovered genes are present on chromosome 6p23-27 in humans.
Diagnosis
Diagnosis is based on the clinical picture including history and EEG findings. The demonstration of Lafora bodies within the apocrine sweat gland of the skin by an axillary skin biopsy examination is a classic approach that may now be substituted by genetic testing. The inclusion bodies, which seem to contain high levels of carbohydrates, are typically labeled by a specific stain called PAS (Periodic acid-Schiff) which is resistant to diastase treatment. Under strong clinical suspicion, liver and brain biopsies may be undertaken. Currently the preferred method of certain diagnosis is DNA sequencing.
Lafora bodies
Lafora disease is distinguished by the presence of inclusions called "Lafora bodies" within the cytoplasm. Lafora bodies are composed of abnormal glycogen called polyglucosans. These amylopectin-like polyglucosans are insoluble and hence precipitate inside cells.
Polyglucosan bodies appear with age; in Lafora disease, their numbers have increased enormously. Lafora bodies have been observed in virtually all organs of patients with the disease. In the brain, their presence appears to be restricted to neurons; they do not seem to present in astrocytes. Their morphology varies from tissue to tissue, but they generally contain a central core and have a peripheral fluffy appearance.[9]
Pathophysiology
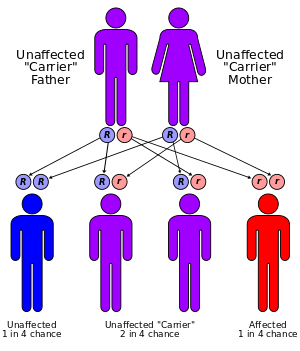
Current understanding of pathophysiology is largely restricted to understanding the generation of Lafora bodies, and their exclusive appearance in neurons and not in astrocytes.
Normal glycogen is soluble in the cellular environment, which has been attributed to its fractal structure. By contrast, the "abnormal glycogen" in Lafora bodies has an excessive phosphate content and branches at abnormally long intervals. It has been shown that laforin dephosphorylates glycogen and preserves its solubility. Hence, in a laforin mutation, glycogen would be hyperphosphorylated. This has been confirmed in laforin knock-out mice.
Research literature suggests that over-activity of glycogen synthase, the key enzyme in synthesizing glycogen, can lead to the formation of polyglucosans. Glycogen synthase can be inactivated by phosphorylation at various amino acid residues by many molecules, including GSK-3beta. Protein phosphatase 1 can take out these phosphate moieties and make glycogen synthase active. However, PP-1 needs other proteins, like PTG (Protein Targeted to Glycogen), to assist. Malin, another protein mutated in Lafora disease, aids in the degradation of PTG, with assistance from laforin via the ubiquitin proteasome system (UPS). Hence in a malin mutation, PTG might accumulate and cause excessive glycogen synthase activity, leading to abnormal glycogen production. However, this supposition has not been confirmed by animal models.
Neurons, though having a capacity to express glycogen synthase, lack capacity to degrade it. They seem not to have glycogen phosphorylase, which is present in astrocytes to degrade glycogen. Astrocytes contribute almost exclusively to brain glycogen storage yet do not develop Lafora bodies, which might highlight the importance of the capacity to degrade glycogen. In a laforin or malin mutation, a laforin–malin complex would cease to exist and drive neurons to make glycogen. This could be detrimental to neuronal function and possibly result in the manifestation of dementia.
Removal of PTG in mice resulted in the near-complete disappearance of polyglucosans and in resolution of neurodegeneration and myoclonic epilepsy.[10]
Prognosis
There is no treatment, and therapy is mainly supportive and symptomatic. Although seizure and myoclonus can be controlled for a long period by using antiepileptic drugs, patients rarely survive beyond one or two decades due to the devastating effects of dementia and ataxia. One medication, Zonisamide, has been shown to lengthen the life of those with the disease. This medication helps control and decrease the severity of the seizures affected patients often experience. Gene therapy strategies are being tried in a mouse model.
Epidemiology
Epilepsy occurs in one percent of all humans. Progressive myoclonic epilepsies (PME) account for about one percent of all epilepsies. Lafora disease is one of the common PMEs. Symptoms of Lafora disease begin to manifest themselves in children from 10 to 17 years old. Males and females are equally affected. There are only 200 reported cases in the world [11] but the exact prevalence is unknown. Most patients affected with this condition have ancestry from the Mediterranean, the Middle East,and India.[12]
History
The disease is named after Gonzalo Rodríguez Lafora (1886–1971), a Spanish neuropathologist who first recognized small inclusion bodies in Lafora patients.[13]
References
- ↑ http://www.rightdiagnosis.com/medical/melf.htm
- 1 2 Ianzano L, Zhang J, Chan EM, Zhao XC, Lohi H, Scherer SW, Minassian BA (2005). "Lafora progressive Myoclonus Epilepsy mutation database - EPM2A and NHLRC1 (EPM2B) genes". Hum Mutat. 26 (4): 397. doi:10.1002/humu.9376. PMID 16134145.
- ↑ James, William D.; Berger, Timothy G. (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. ISBN 0-7216-2921-0.
- ↑ Ortolano S, Vieitez I et al. Loss of cortical neurons underlies the neuropathology of Lafora disease. Mol Brain 2014;7:7 PMCID: PMC3917365
- ↑ Minassan (2000). "Lafora's Disease:Towards a Clinical, Pathologic, and Molecular Synthesis". Pediatr Neurol. 25 (1): 21–29. doi:10.1016/S0887-8994(00)00276-9. PMID 11483392.
- ↑ Minassian, B. A.; Lee, J. R.; Herbrick, J.-A.; Huizenga, J.; Soder, S.; Mungall, A. J.; Dunham, I.; Gardner, R.; Fong, C. G.; Carpenter, S.; Jardim, L.; Satishchandra, P.; Andermann, E.; Snead, O. C., III; Lopes-Cendes, I.; Tsui, L.-C.; Delgado-Escueta, A. V.; Rouleau, G. A.; Scherer, S. W.: Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nature Genet. 20: 171-174, 1998.
- ↑ Chan, E. M.; Bulman, D. E.; Paterson, A. D.; Turnbull, J.; Andermann, E.; Andermann, F.; Rouleau, G. A.; Delgado-Escueta, A. V.; Scherer, S. W.; Minassian, B. A.: Genetic mapping of a new Lafora progressive myoclonus epilepsy locus (EPM2B) on 6p22. J. Med. Genet. 40: 671-675, 2003.
- ↑ (J Neurol Sci 2013;325:170, Brain 2012;135:2684)
- ↑ Striano P, Zara F, Turnbull J, Girard JM, Ackerley CA, Cervasio M, De Rosa G, Del Basso-De Caro L, Striano S, Minassian BA (2008). "Typical Progression of myoclonic epilepsy of the Lafora type". Nat Clin Pract Neurol. 4 (2): 106–111. doi:10.1038/ncpneuro0706. PMID 18256682.
- ↑ Turnbull J, Depaoli-Roach AA, Zhao X, Cortez MA, Pencea N, Tiberia E, Piliguian M, Roach PJ, Wang P, Ackerley CA, Minassian BA (2011) PTG Depletion Removes Lafora Bodies and Rescues the Fatal Epilepsy of Lafora Disease. PLoS Genet 7(4):e1002037
- ↑ "FAMILY TURNS DISEASE INTO SUPPORT FOR OTHERS".
- ↑ "Lafora progressive myoclonus epilepsy".
- ↑ Lafora's disease at Who Named It?
External links
- Lafora disease - OxfordJournals.org
- Chelsea's Hope- Lafora Children's Research Fund
- GeneReview/NCBI/NIH/UW entry on Progressive Myoclonus Epilepsy, Lafora Type
- Support for owners of mini wire dachshunds affected by Lafora