Intramolecular Heck reaction
The intramolecular Heck reaction (IMHR) is the coupling of an aryl or alkenyl halide with an alkene in the same molecule. The reaction may be used to produce carbocyclic or heterocyclic organic compounds with a variety of ring sizes. Chiral palladium complexes can be used to synthesize chiral intramolecular Heck reaction products in non-racemic form.[1]
Introduction
The Heck reaction is the palladium-catalyzed coupling of an aryl or alkenyl halide with an alkene to form a substituted alkene.[2] Intramolecular variants of the reaction may be used to generate cyclic products containing endo or exo double bonds. Ring sizes produced by the intramolecular Heck reaction range from four to twenty-seven atoms. Additionally, in the presence of a chiral palladium catalyst, the intramolecular Heck reaction may be used to establish tertiary or quaternary stereocenters with high enantioselectivity.[3] A number of tandem reactions, in which the intermediate alkylpalladium complex is intercepted either intra- or intermolecularly before β-hydride elimination, have also been developed.[4]
(1)
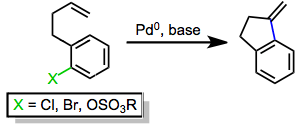
Mechanism and Stereochemistry
The Neutral Pathway
As shown in Eq. 2, the neutral pathway of the Heck reaction begins with the oxidative addition of the aryl or alkenyl halide into a coordinatively unsaturated palladium(0) complex (typically bound to two phosphine ligands) to give complex I. Dissociation of a phosphine ligand followed by association of the alkene yields complex II, and migratory insertion of the alkene into the carbon-palladium bond establishes the key carbon-carbon bond. Insertion takes place in a suprafacial fashion, but the dihedral angle between the alkene and palladium-carbon bond during insertion can vary from 0° to ~90°. After insertion, β-hydride elimination affords the product and a palladium(II)-hydrido complex IV, which is reduced by base back to palladium(0).[5]
(2)
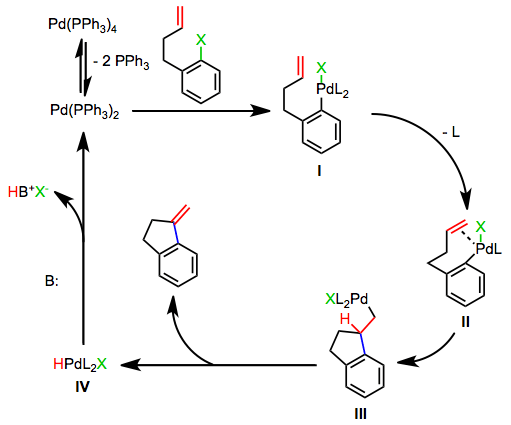
The Cationic Pathway
Most asymmetric Heck reactions employing chiral phosphines proceed by the cationic pathway, which does not require the dissociation of a phosphine ligand. Oxidative addition of an aryl perfluorosulfonate generates a cationic palladium aryl complex V. The mechanism then proceeds as in the neutral case, with the difference that an extra site of coordinative unsaturation exists on palladium throughout the process. Thus, coordination of the alkene does not require ligand dissociation. Stoichiometric amounts of base are still required to reduce the palladium(II)-hydrido complex VIII back to palladium(0).[6] Silver salts may be used to initiate the cationic pathway in reactions of aryl halides.[7]
(3)
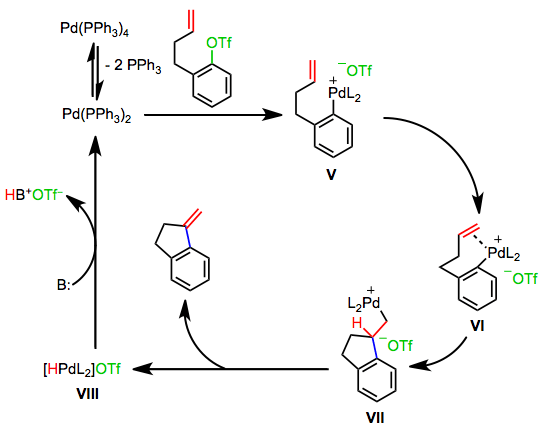
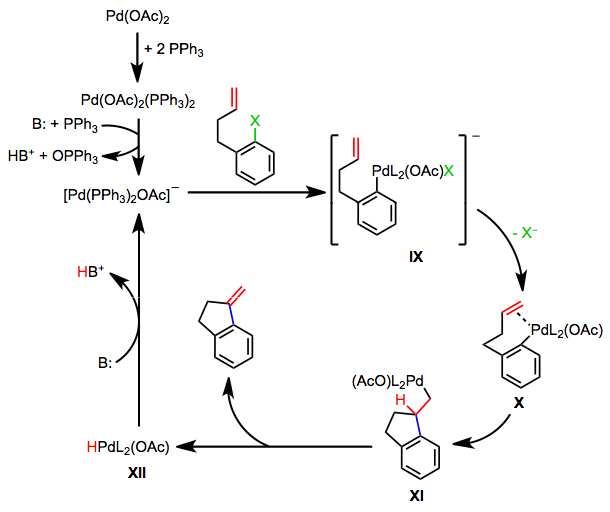
The Anionic Pathway
Reactions involving palladium(II) acetate and phosphine ligands proceed by a third mechanism, the anionic pathway.[8] Base mediates the oxidation of a phosphine ligand by palladium(II) to a phosphine oxide. Oxidative addition then generates the anionic palladium complex IX. Loss of halide leads to neutral complex X, which undergoes steps analogous to the neutral pathway to regenerate anionic complex IX. A similar anionic pathway is also likely operative in reactions of bulky palladium tri(tert-butyl)phosphine complexes.[9]
(4)
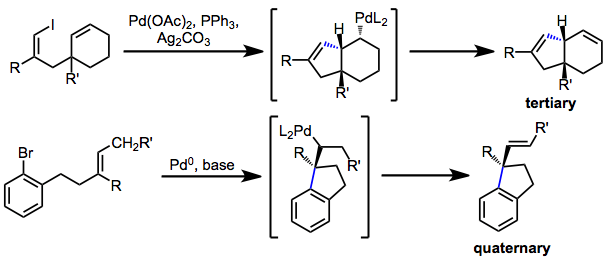
Establishing Tertiary or Quaternary Stereocenters
Asymmetric Heck reactions establish quaternary or tertiary stereocenters. If migratory insertion generates a quaternary center adjacent to the palladium-carbon bond (as in reactions of trisubstituted or 1,1-disubstituted alkenes), β-hydride elimination toward that center is not possible and it is retained in the product.[3] Similarly, β-hydride elimination is not possible if a hydrogen syn to the palladium-carbon bond is not available. Thus, tertiary stereocenters can be established in conformationally restricted systems.[10]
(5)
Scope and Limitations
The intramolecular Heck reaction may be used to form rings of a variety of sizes and topologies. β-Hydride elimination need not be the final step of the reaction, and tandem methods have been developed that involve the interception of palladium alkyl intermediates formed after migratory insertion by an additional reactant. This section discusses the most common ring sizes formed by the intramolecular Heck reaction and some of its tandem and asymmetric variants.
5-Exo cyclization, which establishes a five-membered ring with an exocyclic alkene, is the most facile cyclization mode in intramolecular Heck reactions. In this and many other modes of intramolecular Heck cyclization, annulations typically produce a cis ring juncture.[11]
(6)
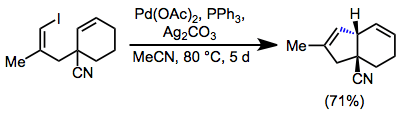
6-Exo cyclization is also common. The high stability of Heck reaction catalysts permits the synthesis of highly strained compounds at elevated temperatures. In the example below, the arene and alkene must both be in energetically unfavorable axial positions in order to react.[12]
(7)

Endo cyclization is observed most often when small or large rings are involved. For instance, 5-endo cyclization is generally preferred over 4-exo cyclization.[13] The yield of endo product increases with increasing ring size in the synthesis of cycloheptenes, -octenes, and -nonenes.[14]
(8)
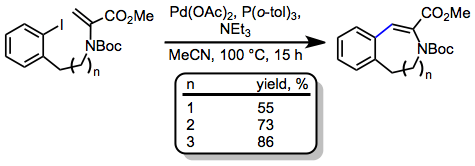
Tandem reactions initiated by IMHR have been extensively explored. Palladium alkyl intermediates generated after migratory insertion may undergo a second round of insertion in the presence of a second alkene (either intra- or intermolecular).[15] When dienes are involved in the intramolecular Heck reaction, insertion affords π-allylpalldium intermediates, which may be intercepted by nucleophiles. This idea was applied to a synthesis of (–)-morphine.[16]
(9)

Asymmetric IMHR may establish tertiary or quaternary stereocenters. BINAP is the most commonly chiral ligand used in this context. An interesting application of IMHR is group-selective desymmetrization (enantiotopic group selection), in which the chiral palladium aryl intermediate undergoes insertion predominantly with one of the enantiotopic double bonds.[17]
(10)
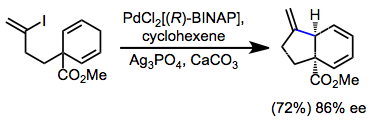
Synthetic Applications
The high functional group tolerance of the intramolecular Heck reaction allows it to be used at a very late stage in synthetic routes. In a synthesis of (±)-FR900482, IMHR establishes a tricyclic ring system in high yield without disturbing any of the sensitive functionality nearby.[18]
(11)
Intramolecular Heck reactions have been employed for the construction of complex natural products. An example is the late-stage, macrocyclic ring closure in the total synthesis of the cytotoxic natural product (–)-Mandelalide A.[19] In another example a fully intramolecular tandem Heck reaction is used in a synthesis of (–)-scopadulcic acid. A 6-exo cyclization sets the quaternary center and provides a neopentyl σ-palladium intermediate, which undergoes a 5-exo reaction to provide the ring system.[20]
(12)

Comparison with Other Methods
The closest competing method to IMHR is radical cyclization.[21] Radical cyclizations are often reductive, which can cause undesired side reactions to occur if sensitive substrates are employed. The IMHR, on the other hand, can be run under reductive conditions if desired.[22] Unlike the IMHR, radical cyclization does not require the coupling of two sp2-hybridized carbons. In some cases, the results of radical cyclization and IMHR are complementary.[23]
Experimental Conditions and Procedure
Typical Conditions
A variety of experimental concerns exist for IMHR reactions. Although most of the common Pd(0) catalysts are commercially available (Pd(PPh3)4, Pd2(dba)3, and derivatives), they may also be prepared by simple, high-yielding procedures.[24] Palladium(II) acetate is cheap and may be reduced in situ to palladium(0) with phosphine. Three equivalents of phosphine per equivalent of palladium acetate are commonly used; these conditions generate Pd(PR3)2 as the active catalyst. Bidentate phosphine ligands are common in asymmetric reactions to enhance stereoselectivity.
A wide variety of bases may be used, and the base is often employed in excess. Potassium carbonate is the most common base employed, and inorganic bases are generally used more often than organic bases. A number of additives have also been identified for the Heck reaction—silver salts may be used to drive the reaction down the cationic pathway, and halide salts may be used to convert aryl triflates via the neutral pathway. Alcohols have been shown to enhance catalyst stability in some cases,[25] and acetate salts are beneficial in reactions following the anionic pathway.[8]
Example Procedure
(13)

A solution of the amide (0.365 g, 0.809 mmol), Pd(PPh3)4 (0.187 g, 0.162 mmol), and triethylamine (1.12 mL, 8.08 mmol) in MeCN (8 mL) in a sealed tube was heated slowly to 120°. After stirring for 40 hours, the reaction mixture was cooled to room temperature, and the solvent was evaporated. The residue was chromatographed (loaded with CH2Cl2) to give the title product 316 (0.270 g, 90%) as a colorless oil; Rf 0.42 (EtOAc/petroleum ether 10:1); [α]22D +14.9 (c, 1.0, CHCl3); IR 3027, 2930, 1712, 1673, 1608, 1492, 1343, 1248 cm−1; 1H NMR (400 MHz) δ 7.33–7.21 (m, 6 H), 7.07 (dd, J = 7.3, 16.4 Hz, 1 H), 7.00 (t, J = 7.5 Hz, 1 H), 6.77 (d, J = 7.7 Hz, 1 H), 6.30 (dd, J = 8.7, 11.4 Hz, 1 H), 5.32 (d, J = 15.7 Hz, 1 H), 5.04 (s, 1 H), 4.95 (s, 1 H), 4.93 (d, J = 11.1 Hz, 1 H), 4.17 (s, 1 H), 3.98 (d, J = 15.7 Hz, 1 H), 3.62 (d, J = 8.7 Hz, 1 H), 3.17 (s, 3 H), 2.56 (dd, J = 3.5, 15.5 Hz, 1 H), 2.06 (dd, J = 2.8, 15.5 Hz, 1 H); 13C NMR (100 MHz) δ 177.4, 172.9, 147.8, 142.2, 136.5, 132.2, 131.6, 128.8, 128.4, 128.2, 127.7, 127.1, 123.7, 122.9, 107.9, 105.9, 61.0, 54.7, 49.9, 44.4, 38.2, 26.4; HRMS Calcd. for C24H22N2O2: 370.1681. Found: 370.1692.
References
- ↑ Link, J. T. Org. React. 2002, 60, 157. doi:10.1002/0471264180.or060.02
- ↑ Beletskaya, I.; Cheprakov, A. Chem. Rev. 2000 100, 3009.
- 1 2 Overman, L. E. Pure Appl. Chem. 1994, 66, 1423.
- ↑ Larock, R. C.; Lee, N. H. J. Org. Chem. 1991, 56, 6253.
- ↑ Amatore, C.; Azzabi, M.; Jutand, A. J. Am. Chem. Soc. 1991, 113, 8375.
- ↑ Cabri, W.; Candiani, I.; DeBernardinis, S.; Francalanci, F.; Penco, S.; Santi, R. J. Org. Chem. 1991, 56, 5796.
- ↑ Karabelas, K.; Westerlund, C.; Hallberg, A. J. Org. Chem. 1985, 50, 3896.
- 1 2 Amatore, C.; Jutand, A. Acc. Chem. Res. 2000, 33, 314.
- ↑ Carrow, B.; Hartwig, J. F. J. Am. Chem. Soc., 2010, 132, 79.
- ↑ Honzawa, H.; Mizutani, T.; Shibasaki, M. Tetrahedron Lett. 1999, 40, 311.
- ↑ Larock, R. C.; Song, H.; Baker, B. E.; Gong, W. H. Tetrahedron Lett. 1988, 29, 2919.
- ↑ Wünsch, B.; Diekmann, H.; Höfner, G. Tetrahedron: Asymmetry 1995, 6, 1527.
- ↑ Koerber-Plé, K.; Massiott, G. Synlett 1994, 759.
- ↑ Gibson, S. E.; Guillo, N.; Middleton, R. J.; Thuillez, A.; Tozer, M. J. J. Chem. Soc., Perkin Trans. 1 1997, 447.
- ↑ Grigg, R.; Sridharan, V. Tetrahedron Lett. 1992, 33, 7965.
- ↑ Hong, C. Y.; Overman, L. E. Tetrahedron Lett. 1994, 35, 3453.
- ↑ Sato, Y.; Mori, M.; Shibasaki, M. Tetrahedron Lett. 1992, 33, 2593.
- ↑ Schkeryantz, J. M.; Danishefsky, S. J. J. Am. Chem. Soc. 1995, 117, 4722.
- ↑ Nguyen, M. H.; Imanishi, M.; Kurogi, T.; Smith, A. B. III J. Am. Chem. Soc. 2016, 138, 3675.
- ↑ Fox, M. E.; Li, C.; Marino, J. P.; Overman, L. E. J. Am. Chem. Soc. 1999, 121, 5467.
- ↑ Giese, B.; Kopping, B.; Göbel, T.; Dickhaut, J.; Thoma, G.; Kulicke, K. j.; Trach, F. Org. React. 1997, 48, 301.
- ↑ Burns, B.; Grigg, R.; Ratananukul, P.; Sridharan, V.; Stevenson, P.; Worakun, T. Tetrahedron Lett. 1988, 29, 4329.
- ↑ Ishibashi, H.; Ito, K.; Hirano, T.; Tabuchi, M.; Ikeda, M. Tetrahedron 1993, 49, 4173.
- ↑ Hegedus, L. S. In Organometallics in Synthesis; Schlosser, M., Ed.; John Wiley and Sons Ltd.: Chichester, England, 1994; p. 383.
- ↑ Ohrai, K.; Kondo, K.; Sodeoka, M.; Shibasaki, M. J. Am. Chem. Soc. 1994, 116, 11737.