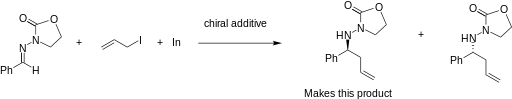
Indium-mediated allylation
Indium-Mediated Allylations (IMAs) are important chemical reactions for the formation of carbon–carbon bonds. This reaction has two steps: first, indium inserts itself between the carbon–halogen bond of an allyl halide, becoming the organoindium intermediate; second, this allyl indide intermediate reacts with an electrophile to synthesize one of a wide range of compounds, such as carbohydrates and antihelminthic drugs. This reaction is depicted in the scheme below:

Although this reaction occurs in two steps, it is commonly done as a Barbier reaction where the indium, allyl halide, and electrophile are all mixed together in a one-pot process.

Indium reacts more readily than other metals, such as Mg, Pb, Bi, or Zn and does not require a promoter or flammable organic solvent to drive the reaction. IMAs have advantages over other carbon bond forming reactions because of their ability to be carried out in water, which is cheap and environmentally friendly. Therefore, these reactions represent Green chemistry, providing a safer alternative to the very common Grignard reaction, performed with Mg. Reactions yield high stereo- and regio-selectivity with few by-products making it easy to purify the desired product.
Industrial use
The ability of the IMA reaction to form new carbon–carbon bonds allows simple molecules found in petroleum to be built into bigger, more complex molecules like drugs. Indium metal is very good at selectively synthesizing specific products.
Reaction conditions
In reactions with indium, water is widely used as solvent. Reactions with magnesium, in contrast, must exclude water since protonolysis destroys the Grignard reagent. Furthermore, Mg is often coated with an oxide layer of MgO, requiring heat, sonication, or chemical treatment to expose fresh metal. Indium, however, can readily react with allyl halides in water. After the intermediate reacts with the electrophile, free In3+ ions are released which may rapidly precipitate as In(OH)3. Under some conditions, this precipitate may coat the surface of the indium and hinder the reaction. To prevent this side reaction from complicating the synthesis, an acid such as HCl or acetic acid is often added.
Other solvents
Although indium mediated allylations can be carried out in aqueous media, a variety of other solvents may be used including THF (tetrahydrofuran), DMF (dimethylformamide), room temperature ionic liquids, NMF (n-methylformamide), and others. [1] [2] Solvent often affects the solubility, rate of the reaction, yield, stability, regioselectivity, and stereoselectivity.
Diversity of reaction
The organic halide
Allyl bromides and iodides are common reactants for the IMA reaction. Chlorides are also used but are likely to proceed more slowly since the carbon–chlorine bond is relatively strong. Fluorides are generally unreactive, and thus not used.
Further increasing the importance of indium, propargyl halides undergo similar reactions. An example is shown below.
![]()
Reactions with electrophilic compounds
Indium mediates the allylation of a wide variety of electrophiles. The examples in the following scheme illustrate the breadth of applications of IMA.

Selectivity
Indium mediated allylations are very selective, offering four different types of selectivity: chemoselectivity, regioselectivity, diastereoselectivity, and enantioselectivity.
Chemoselective chemistry
Organoindium intermediates do not react with –OH or –CO2H groups. Reactions with carbonyls, however, give high yields. Research has shown that in reactions of an indium intermediate with an electrophilic compound of both aldehyde and ketone, the reaction proceeded with the aldehyde. The electrophilic compound is shown below.[3]

Regioselective chemistry
The regioselectivity of allylation mediated by indium in water is dependent on the steric effects of the substituents on both the intermediate and carbonyl. An α-attack from the nucleophile (at the position bearing the halogen) is distinguishable from a γ-attack (at the double bond) by inspecting the products. The scheme below gives an example of two different products formed from the same nucleophile under α-regioselectivity (α) and γ-regioselectivity (γ). This regioselectivity does not appear to depend on conjugation or the degree of substitution. [4]

Diastereoselective chemistry
The addition of allylindium reagents to aldehydes substituted at α or β carbons can be very diastereoselective in aqueous systems. For example, if chelation control is present in an α-oxy aldehyde, the product is expected to be the syn diastereomer. A sample reaction of chelation versus non-chelation control is illustrated below.

Numerous investigations have found an explanation for this effect. The oxygens of the carbonyl and the hydroxyl group chelate the indium of the organoindium intermediate as illustrated below on the left by the two green bonds. The incipient C-C bond, illustrated in red, creates a six-member ring in a chair conformation. Under chelation control, the allyl group attacks the carbonyl carbon from the less hindered side opposite to that of the R group. Once the C-C bond is fully formed, the indium is released, producing the syn diol. A similar chelated structure explains why allylation of β-oxy aldehydes results in anti diols.[5]

From this application of allylindium and oxygenated aldehydes, pharmaceutical companies can select for specific diastereomeric products. The Grignard reaction does not have this advantage because it is so reactive that it attacks indiscriminately from both sides.
Enantioselective chemistry
Indium is also capable of affording excellent enantioselectivity when performed in the presence of chiral additives. For example, three effective chiral additives are shown below.
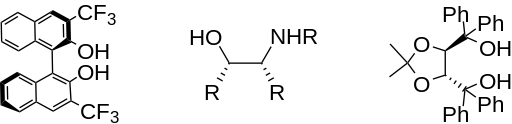
The addition of allylindium reagents to electrophilic hydrazones, illustrated below, has been reported to synthesize only one enantiomer of the chiral product with up to 97% selectivity using binol as a chiral additive. [6] Similarly, a chiral amino alcohol allows for extremely high enantioselectivity in the allylation of ketones. [7]

Fundamental studies of IMA
Organoindium intermediate
Although there has been some confusion over the precise structure of the organoindium intermediates, recent publications have added considerable clarity. [8] For example, after mixing allyl bromide with indium in THF, two compounds were separately crystallized and identified as monoallylindium dibromide and diallylindium bromide, illustrated below. [9]

However, other species may also be present. In solution, mass spectrometry has detected a variety of organoindium(III) species such as InRX3−, RXIn+, R2In+, X2In+, many of which are in equilibrium with each other. [10]
Balanced equation
Although most attention is paid to the organic molecules produced by these reactions, the overall balanced equation of IMA has been determined in aqueous media. The formation of the organoindium intermediates is illustrated in the first reaction below. Inclusion of, for example, an aldehyde as the electrophile is represented by the following reaction. (The proportion of indium halide to indium hydroxide will depend on conditions such as pH and temperature.)

Rate of reaction
The rate of this reaction, determined approximately by TLC, appears to be solvent dependent. The first step, formation of the organoinidum intermediate at the indium surface, is rapid and first order with respect to allyl halide. When the halide is bromide or iodide and convection is rapid, the reaction rate is mass transport limited. The heterogeneous rate constant for the first step has been determined to be at least 0.01 cm/s. [11]
Thermodynamics
Formation of organoindium intermediates is known to be exothermic, but no measurements of the enthalpy or free energy of the reactions have been reported.
References
- ↑ Frimpong, K; Wzorek, J; Lawlor, C; Spencer, K; Mitzel. T; J. Org. Chem. 2009, 74, 5861–5870. doi:10.1021/jo900763u
- ↑ Law, M.C; Cheung, T.W; Wong, K.Y; Chan, T.H. J. Org. Chem. 2007, 72, 923–929.
- ↑ Haddad, T.D; Hirayama, L.C; Buckley, J.J; Singaram, B. J. Org. Chem. 2012, 77, 889–898.
- ↑ Isaac, M.B; Chan, T.H. Tetrahedron Lett. 1995, 36, 8957–8960.
- ↑ Paquette, L.A; Mitzel, T.M. J. Am. Chem. Soc. 1996, 118, 1931–1937.
- ↑ Cook, G.R; Kargbo, R; Maity, B. Org. Lett. 2005, 7, 2767–2770.
- ↑ Haddad, T.D; Hirayama, L.C; Taynton, P; Singaram, B. Tetrahedron Lett. 2008, 49, 508–511.
- ↑ Bowyer, W.J; Singaram, B; Sessler, A.M. Tetrahedron 2011, 67, 7449–7460.
- ↑ Yasuda, M; Haga, M; Nagaoka, Y; Baba, A. Eur. J. Org. Chem. 2010, 5359–5363.
- ↑ Koszinowski, K. J. Am. Chem. Soc. 2010, 132, 6032–6040.
- ↑ Olson, I.A.; Bacon, W.A.; Baez Sosa, Y.Y.; Delaney, K.M.; Forte, S.A.; Guglielmo, M.A.; Hill, A.N.; Kiesow, K.H.; Langenbacher, R.E.; Xun, Y.; Young, R.O.; Bowyer, W.J. J. Phys. Chem. A 2011, 115, 11001-11007.