Hemophagocytic lymphohistiocytosis
| Hemophagocytic lymphohistiocytosis | |
|---|---|
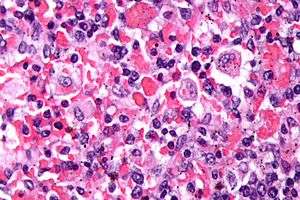 | |
| Micrograph showing red blood cells within macrophages. H&E stain. | |
| Classification and external resources | |
| Specialty | hematology |
| ICD-10 | D76.1 |
| ICD-9-CM | 288.4 |
| OMIM | 267700 603553 608898 603552 |
| DiseasesDB | 31418 |
| eMedicine | ped/745 |
| MeSH | D051359 |
| GeneReviews | |
Hemophagocytic lymphohistiocytosis (HLH), also known as haemophagocytic lymphohistiocytosis (British spelling), and hemophagocytic or haemophagocytic syndrome,[1] is an uncommon hematologic disorder. It is a life-threatening disease of severe hyperinflammation caused by uncontrolled proliferation of activated lymphocytes and macrophages, characterised by proliferation of morphologically benign lymphocytes and macrophages that secrete high amounts of inflammatory cytokines. It is classified as one of the cytokine storm syndromes.
History
The first case report of HLH was published in 1952.[2]
Classification
Primary HLH, also known as familial haemophagocytic lymphohistiocytosis (FHL) or familial erythrophagocytic lymphohistiocytosis, is a heterogeneous autosomal recessive disorder found to be more prevalent with parental consanguinity.
Secondary haemophagocytic lymphohistiocytosis (acquired haemophagocytic lymphohistiocytosis) occurs after strong immunologic activation, such as that which can occur with systemic infection, immunodeficiency, or underlying malignancy.
Both forms are characterized by the overwhelming activation of normal T lymphocytes and macrophages, invariably leading to clinical and haematologic alterations and death in the absence of treatment.
Genetics
Five genetic subtypes (FHL1, FHL2, FHL3, FHL4, and FHL5) are described, with an estimated prevalence of one in 50,000 and equal gender distribution. Molecular genetic testing for four of the causative genes, PRF1 (FHL2), UNC13D (FHL3), STX11 (FHL4), and STXBP2 (FHL5), is available on a clinical basis. Symptoms of FHL are usually evident within the first few months of life and may even develop in utero. However, symptomatic presentation throughout childhood and even into young adulthood has been observed in some cases.
The five subtypes of FHL[3] are each associated with a specific gene:
- FHL1: HPLH1
- FHL2: PRF1 (Perforin)
- FHL3: UNC13D (Munc13-4)
- FHL4: STX11 (Syntaxin 11)
- FHL5: STXBP2 (Syntaxin binding protein 2)/UNC18-2
Nearly half of the cases of type 2 familial hemophagocytic lymphohistiocytosis are due to bi-allelic PRF1 mutations.[4]
Clinical presentation
The onset of HLH occurs under the age of 1 year in ~70% of cases. Familial HLH should be suspected if siblings are diagnosed with HLH or if symptoms recur when therapy has been stopped. Each full sibling of a child with familial HLH has a 25% chance of developing the disease, a 50% chance of carrying the defective gene (which is very rarely associated with any risk of disease) and a 25% chance of not being affected and not carrying the gene defect.
Patient with HLH, especially when untreated, may need intensive therapy. Therefore, HLH should be included in the differential diagnosis of ICU (Intensive Care Unit) patients with cytopenia and hyperferritinemia.[5]
HLH clinically manifests with fever, hepatosplenomegaly, lymphadenopathy, jaundice and a rash.
Investigations

The blood count typically shows pancytopenia—anemia, neutropenia, and thrombocytopenia.
The bone marrow may show hemophagocytosis.
The liver function tests are usually elevated. Hypoalbuminemia is common.
The serum C reactive protein, erythrocyte sedimentation rate and ferritin level are markedly elevated. In pediatric populations a ferritin above 10000 is very sensitive and specific for the diagnosis of HLH,[6] however, the diagnostic utility for ferritin is less for adult HLH patients.[7]
The serum fibrinogen level is usually low and the D-dimer level is elevated.
The sphingomyelinase is elevated.[8]
Bone marrow biopsy shows histiocytosis.[9]
Diagnostic criteria
The current (2008) diagnostic criteria for HLH are[10]
1. A molecular diagnosis consistent with HLH. These include the identification of pathologic mutations of PRF1, UNC13D, or STX11.
OR
2. Fulfillment of five out of the eight criteria below:
- Fever (>100.4 °F, >38 °C)
- Splenomegaly
- Cytopenias affecting at least two of three lineages in the peripheral blood:
- Haemoglobin <9 g/100 ml (in infants <4 weeks: haemoglobin <10 g/100 ml)
- Platelets <100×109/L
- Neutrophils <1×109/L
- Hypertriglyceridemia (fasting, greater than or equal to 265 mg/100 ml) and/or hypofibrinogenemia (≤ 150 mg/100 ml)
- Ferritin ≥ 500 ng/ml
- Haemophagocytosis in the bone marrow, spleen or lymph nodes
- Low or absent natural killer cell activity
- Soluble CD25 (soluble IL-2 receptor) >2400 U/ml (or per local reference laboratory)
In addition, in the case of familial HLH, no evidence of malignancy should be apparent.
It should be noted that not all five out of eight criteria are required for diagnosis of HLH in adults, and a high index of suspicion is required for diagnosis as delays results in increased mortality. The diagnostic criteria were developed in pediatric populations and have not been validated for adult HLH patients.[11] Attempts to improve diagnosis of HLH have included use of the HScore, which can be used to estimate an individual's risk of HLH.[12]
Differential diagnosis
The differential diagnosis of HLH includes secondary HLH and macrophage-activation syndrome or other primary immunodeficiencies that present with hemophagocytic lymphohistiocytosis, such as X-linked lymphoproliferative disease.
Other conditions that may be confused with this condition include autoimmune lymphoproliferative syndrome.[13]
The diagnosis of acquired, or secondary, HLH is usually made in association with infection by viruses, bacteria, fungi, or parasites or in association with lymphoma, autoimmune disease, or metabolic disease. Acquired HLH may have decreased, normal, or increased NK cell activity.
Griscelli syndrome
A major differential of HLH is Griscelli syndrome (type 2). This is a rare (less than 100 reported cases) autosomal recessive disorder characterized by partial albinism, hepatosplenomegaly, pancytopenia, hepatitis, immunologic abnormalities, and lymphohistiocytosis. Most cases have been diagnosed between 4 months and 7 years of age, with a mean age of about 17 months.
Three types of Griscelli syndrome are recognised: Type 1 have neurologic symptoms and mutations in MYO5A. Prognosis depends on the severity of neurologic manifestations. Type 2 have mutations in RAB27A and haemophagocytic syndrome, with abnormal T-cell and macrophage activation. This type has a grave prognosis if untreated. Type 3 have mutations in melanophilin and are characterized by partial albinism. This type does not pose a threat to those so affected.
Treatment
In secondary cases treatment of the cause, where possible, is indicated. Additionally treatment for HLH itself is usually required.
While optimal treatment of HLH is still being debated, current treatment regimes usually involve high dose corticosteroids, etoposide and cyclosporin. Intravenous immunoglobulin is also used. Methotrexate and vincristine have also been used. Other medications include cytokine targeted therapy.
Prognosis
The prognosis is guarded with an overall mortality of 50%. Poor prognostic factors included HLH associated with malignancy, with half the patients dying by 1.4 months compared to 22.8 months for non-tumour associated HLH patients.[14]
Secondary HLH in some individuals may be self-limited because patients are able to fully recover after having received only supportive medical treatment (i.e., IV immunoglobulin only). However, long-term remission without the use of cytotoxic and immune-suppressive therapies is unlikely in the majority of adults with HLH and in those with involvement of the central nervous system (brain and/or spinal cord).[3]
See also
References
- ↑ Fisman, David N. (2000). "Hemophagocytic syndromes and infection". Emerging Infect. Dis. 6 (6): 601–8. doi:10.3201/eid0606.000608. PMC 2640913
 . PMID 11076718.
. PMID 11076718. - ↑ Farquhar, James W.; Claireaux, Albert E. (December 1952). "Familial Haemophagocytic Reticulosis". Archives of Disease in Childhood. 27 (136): 519–525. doi:10.1136/adc.27.136.519. PMC 1988563. PMID 13008468.
- 1 2 Zhang, Kejian; Filopovich, Alexandra H.; Johnson, Judith; Marsh, Rebecca A.; Villanueva, Joyce (January 17, 2013). "Hemophagocytic Lymphohistiocytosis, Familial". GeneReviews. National Center for Biotechnology Information. PMID 20301617. NBK1444.
- ↑ Trapani JA, Thia KY, Andrews M, et al. (April 2013). "Human perforin mutations and susceptibility to multiple primary cancers". Oncoimmunology. 2 (4): e24185. doi:10.4161/onci.24185. PMC 3654607. PMID 23734337.
- ↑ Machowicz, Rafal; Janka, Gritta; Wiktor-Jedrzejczak, Wieslaw (2016-01-01). "Your critical care patient may have HLH (hemophagocytic lymphohistiocytosis)". Critical Care. 20: 215. doi:10.1186/s13054-016-1369-3. ISSN 1364-8535. PMC 4937543. PMID 27389585.
- ↑ Allen, Carl (June 2008). "Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis.". Pediatric Blood & Cancer. 50: 1227–35. doi:10.1002/pbc.21423. PMID 18085676.
- ↑ Schram, Alison (March 5, 2015). "Marked hyperferritinemia does not predict for HLH in the adult population". Blood. 125: 1548–52. doi:10.1182/blood-2014-10-602607. PMID 25573993. Retrieved December 14, 2015.
- ↑ Jenkins RW, Clarke CJ, Lucas JT, et al. (November 2013). "Evaluation of the role of secretory sphingomyelinase and bioactive sphingolipids as biomarkers in hemophagocytic lymphohistiocytosis". Am. J. Hematol. 88 (11): E265–72. doi:10.1002/ajh.23535. PMID 23828274.
- ↑ Lymphohistiocytosis, Hemophagocytic at the US National Library of Medicine Medical Subject Headings (MeSH)
- ↑ Jordan MB, Filipovich AH (October 2008). "Hematopoietic cell transplantation for hemophagocytic lymphohistiocytosis: a journey of a thousand miles begins with a single (big) step". Bone Marrow Transplant. 42 (7): 433–7. doi:10.1038/bmt.2008.232. PMID 18679369.
- ↑ Schram, Alison (May 7, 2015). "How I treat hemophagocytic lymphohistiocytosis in the adult patient.". Blood. 125: 2908–14. doi:10.1182/blood-2015-01-551622. PMID 25758828. Retrieved December 14, 2015.
- ↑ Fardet, Laurence (September 9, 2014). "Development and Validation of the HScore, a Score for the Diagnosis of Reactive Hemophagocytic Syndrome". Arthritis & Rheumatology. 66: 2613–20. doi:10.1002/art.38690. PMID 24782338. Retrieved December 14, 2015.
- ↑ Rudman Spergel A, Walkovich K, Price S, et al. (November 2013). "Autoimmune lymphoproliferative syndrome misdiagnosed as hemophagocytic lymphohistiocytosis". Pediatrics. 132 (5): e1440–4. doi:10.1542/peds.2012-2748. PMID 24101757.
- ↑ Parikh, Sameer (April 2014). "Prognostic factors and outcomes of adults with hemophagocytic lymphohistiocytosis.". Mayo Clinic Proceedings. 89: 484–92. doi:10.1016/j.mayocp.2013.12.012. PMID 24581757. Retrieved December 14, 2015.