Fluorescence in the life sciences
Fluorescence is used in the life sciences generally as a non-destructive way of tracking or analysing biological molecules by means of fluorescence. Some proteins or small molecules in cells are naturally fluorescent, which is called intrinsic fluorescence or autofluorescence (such as NADH, tryptophan or endogenous Chlorophyll, Phycoerythrin or green fluorescent protein). Alternatively, specific or general proteins, nucleic acids, lipids or small molecules can be "labelled" with an extrinsic fluorophore, a fluorescent dye which can be a small molecule, protein or quantum dot. Several techniques exist to exploit additional properties of fluorophores, such as fluorescence resonance energy transfer, where the energy is passed non-radiatively to a particular neighbouring dye, allowing proximity or protein activation to be detected; another is the change in properties, such as intensity, of certain dyes depending on their environment allowing their use in structural studies.[1][2]
Fluorescence
.ogg.jpg)
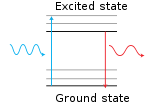
The principle behind fluorescence is that the fluorescent moiety contains electrons which can absorb a photon and briefly enter an excited state before either dispersing the energy non-radiatively or emitting it as a photon, but with a lower energy, i.e., at a longer wavelength (wavelength and energy are inversely proportional).[3] The difference in the excitation and emission wavelengths is called the Stokes shift, and the time that an excited electron takes to emit the photon is called a lifetime. The quantum yield is an indicator of the efficiency of the dye (it is the ratio of emitted photons per absorbed photon), and the extinction coefficient is the amount of light that can be absorbed by a fluorophore. Both the quantum yield and extinction coefficient are specific for each fluorophore and multiplied together calculates the brightness of the fluorescent molecule.[4]
Labelling
Reactive dyes
Fluorophores can be attached to proteins via specific functional groups, such as
- amino groups (e.g. via succinimide, Isothiocyanate or hydrazine)
- carboxyl groups (e.g. via carbodiimide)
- thiol (e.g. via maleimide or acetyl bromide)
- azide (e.g. via click chemistry)
or non-specificately (glutaraldehyde) or non-covalently (e.g. via hydrophobicity, etc.).
These fluorophores are either small molecules, protein or quantum dots.
Organic fluorophores fluoresce thanks to delocalized electrons which can jump a band and stabilize the energy absorbed, hence most fluorophores are conjugated systems. Several families exits and their excitations range from the infrared to the ultraviolet.
Lanthanides (chelated) are uniquely fluorescent metals, which emit thanks to transitions involving 4f orbits, which are forbidden, hence they have very low absorption coefficients and slow emissions, requiring exitation through fluorescent organic chelators (e.g. dipicolinate-based Terbium (III) chelators [5]).
A third class of small molecule fluorophore is that of the transition metal-ligand complexes, which display molecular fluorescence from a metal-to-ligand charge transfer state which is partially forbidden, these are generally complexes of Ruthenium, Rhenium or Osmium.
Quantum dots
Quantum dots are fluorescent semiconductor nanoparticles.
Fluorescent Proteins
Several fluorescent protein exist in nature, but the most important one as a research tool is Green Fluorescent Protein (GFP) from the jellyfish Aequorea victoria,[6] which spontaneously fluoresces upon folding via specific serine-tyrosine-glycine residues. The benefit that GFP and other fluorescent proteins have over organic dyes or quantum dots is that they can be expressed exogenously in cells alone or as a fusion protein, a protein that is created by ligating the fluorescent gene (e.g., GFP) to another gene and whose expression is driven by a housekeeping gene promoter or another specific promoter. This approach allows fluorescent proteins to be used as reporters for any number of biological events, such as sub-cellular localization and expression patterns. A variant of GFP is naturally found in corals, specifically the Anthozoa, and several mutants have been created to span the visible spectra and fluoresce longer and more stably. Other proteins are fluorescent but require a fluorophore cofactor, and hence can only be used in vitro; these are often found in plants and algae (phytofluors, phycobiliprotein such as allophycocyanin).
Bioluminescence and fluorescence
Fluorescence, chemiluminescence and phosphorescence are 3 different types of luminescence properties, i.e. emission of light from a substance. Fluorescence is a property where light is absorbed and remitted within a few nanoseconds (approx. 10ns) at a lower energy (=higher wavelength), while bioluminescence is biological chemiluminescence, a property where light is generated by a chemical reaction of an enzyme on a substrate. Phosphorescence is a property of materials to absorb light and emit the energy several milliseconds or more later (due to forbidden transitions to the ground state of a triplet state, while fluorescence occurs in exited singlet states). Until recently was not applicable to life science research due to the size of the inorganic particles. However the boundary between the fluorescence and phosphorescence is not clean cut as transition metal-ligand complexes, which combine a metal and several organic moieties, have long lifetimes, up to several microseconds (as they display mixed singlet-triplet states).
Comparison with radioactivity
Prior to its widespread use in the past three decades radioactivity was the most common label.
The advantages of fluorescence over radioactive labels are as follows:
- Fluorescence is much safer and more convenient to use
- Several fluorescent molecules can be used simultaneously given that they do not overlap, cf. FRET, whereas with radioactivity two isotopes can be used (tritium and a low energy isotope such as 33P due to different intensities) but require special machinery (a tritium screen and a regular phosphor-imaging screen or a specific dual channel detector)[7]).
- Several properties are extremely useful (cf. next section)
Note: a channel is similar to "colour" but distinct, it is the pair of excitation and emission filters specific for a dye, e.g. agilent microarrays are dual channel, working on cy3 and cy5, these are colloquially referred to as green and red.
Disadvantages of fluorophores include:
- Toxicity
- Interference with normal biological processes
Additional useful properties
The basic property of fluorescence are extensively used, such as a marker of labelled components in cells (fluorescence microscopy) or as an indicator in solution (Fluorescence spectroscopy), but other additional properties, not found with radioactivity, make it even more extensively used.
FRET
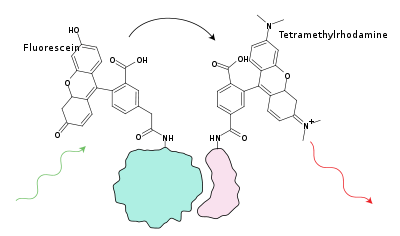
FRET (Förster resonance energy transfer) is a property in which the energy of the excited electron of one fluorphore, called the donor, is passed on to a nearby acceptor dye, either a dark quencher or another fluorophore, which has an excitation spectrum which overlaps with the emission spectrum of the donor dye resulting in a reduced fluorescence. This can be used to
- detect if two labelled protein or nucleic acids come into contact or a doubly labelled single molecules is hydrolysed
- detect changed in conformation
- measure concentration by a competitive binding assay
Sensitivity to environment
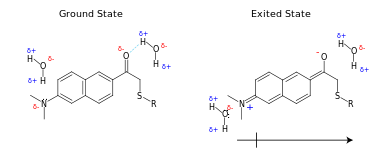
Environment-sensitive dyes change their properties (intensity, half-life, and excitation and emission spectra) depending on the polarity (hydrophobicity and charge) of their environments. examples include: Indole, Cascade Yellow, prodan, Dansyl, Dapoxyl, NBD, PyMPO, Pyrene and diethylaminocumarin.
This change is most pronounced when electron-donating and electron-withdrawing groups are placed at opposite ends of an aromatic ring system,[8] as this results in a large change in dipole moment when excited.
When a fluorophore is excited, it generally has a larger dipole moment (μE) than in the ground state (μG). Absorption of a photon by a fluorophore takes a few picoseconds. Before this energy is released (emission: 1–10 ns), the solvent molecules surrounding the fluorophore reorient (10–100 ps) due to the change in polarity in the excited singlet state; this process is called solvent relaxation. As a result of this relaxation, the energy of the excited state of the fluorophore is lowered (longer wavelength), hence fluorophores that have a large change in dipole moment have larger stokes shift changes in different solvents. The difference between the energy levels can be roughly determined with the Lipper-Mataga equation.
It should be noted that a hydrophobic dye is a dye which is insoluble in water, a property independent of solvatochromism.
Additionally, The term environment-sensitive in chemistry actually describes changes due to one of a variety of different environmental factors, such as pH or temperature, not just polarity; however, in biochemistry environment-sensitive fluorphore and solvatochromic fluorophore are used interchangeably: this convention is so widespread that suppliers describe them as environment-sensitive over solvatochromic.
Fluorescence lifetime
Fluorescent moieties emit photons several nanoseconds after absorption following an exponential decay curve, which differs between dyes and depends on the surrounding solvent. When the dye is attached to a macromolecules the decay curve becomes multiexponential. Conjugated dyes generally have a lifetime between 1–10 ns, a small amount of longer lived exceptions exist, notably pyrene with a lifetime of 400ns in degassed solvents or 100ns in lipids and coronene with 200ns. On a different category of fluorphores are the fluorescent organometals (lanthanides and transition metal-ligand complexes) which have been previously described, which have much longer lifetimes due to the restricted states: lanthanides have lifetimes of 0.5 to 3 ms, while transition metal-ligand complexes have lifetimes of 10 ns to 10 µs. Note that fluorescent lifetime should not be confused with the photodestruction lifetime or the "shelf-life" of a dye.
Multiphoton excitation
Multiphoton excitation is a way of focusing the viewing plane of the microscope by taking advantage of the phenomenon where two simultaneous low energy photons are absorbed by a fluorescent moiety which normally absorbs one photon with double their individual energy: say two NIR photons (800 nm) to excite a UV dye (400 nm).
Fluorescence anisotropy
A perfectly immobile fluorescent moiety when exited with polarized light will emit light which is also polarized. However if a molecule is moving, it will tend to "scramble" the polarization of the light by radiating at a different direction from the incident light.
Methods
- Fluorescence microscopy of tissues, cells or subcellular structures is accomplished by labeling an antibody with a fluorophore and allowing the antibody to find its target antigen within the sample. Labeling multiple antibodies with different fluorophores allows visualization of multiple targets within a single image.
- Automated sequencing of DNA by the chain termination method; each of four different chain terminating bases has its own specific fluorescent tag. As the labeled DNA molecules are separated, the fluorescent label is excited by a UV source, and the identity of the base terminating the molecule is identified by the wavelength of the emitted light.

- DNA detection: the compound ethidium bromide, when free to change its conformation in solution, has very little fluorescence. Ethidium bromide's fluorescence is greatly enhanced when it binds to DNA, so this compound is very useful in visualising the location of DNA fragments in agarose gel electrophoresis. Ethidium bromide can be toxic – a purportedly safer alternative is the dye SYBR Green.
- The DNA microarray
- Immunology: An antibody has a fluorescent chemical group attached, and the sites (e.g., on a microscopic specimen) where the antibody has bound can be seen, and even quantified, by the fluorescence.
- FACS (fluorescent-activated cell sorting)
- Microscale Thermophoresis (MST) uses fluorescence as readout to quantify the directed movement of biomolecules in microscopic temperature gradients.
- Fluorescence has been used to study the structure and conformations of DNA and proteins with techniques such as Fluorescence resonance energy transfer, which measures distance at the angstrom level. This is especially important in complexes of multiple biomolecules.
- Fluorescence can be applied to study colocalization of various proteins of interest.[9] It then can be analyzed using a specialized software, like CoLocalizer Pro.
Also, many biological molecules have an intrinsic fluorescence that can sometimes be used without the need to attach a chemical tag. Sometimes this intrinsic fluorescence changes when the molecule is in a specific environment, so the distribution or binding of the molecule can be measured. Bilirubin, for instance, is highly fluorescent when bound to a specific site on serum albumin. Zinc protoporphyrin, formed in developing red blood cells instead of hemoglobin when iron is unavailable or lead is present, has a bright fluorescence and can be used to detect these problems.
The number of fluorescence applications in the biomedical, biological and related sciences continuously expands. Methods of analysis in these fields are also growing, often with nomenclature in the form of acronyms such as: FLIM, FLI, FLIP, CALI, FLIE, FRET, FRAP, FCS, PFRAP, smFRET, FIONA, FRIPS, SHREK, SHRIMP or TIRF. Most of these techniques rely on fluorescence microscopes, which use high intensity light sources, usually mercury or xenon lamps, LEDs, or lasers, to excite fluorescence in the samples under observation. Optical filters then separate excitation light from emitted fluorescence to be detected by eye or with a (CCD) camera or other light detector (e.g., photomultiplier tubes, spectrographs). Considerable research is underway to improve the capabilities of such microscopes, the fluorescent probes used, and the applications they are applied to. Of particular note are confocal microscopes, which use a pinhole to achieve optical sectioning, which affords a quantitative, 3D view of the sample.
See also
References
- ↑ Joseph R. Lakowicz (2006). Principles of fluorescence spectroscopy. Springer. pp. 26–. ISBN 978-0-387-31278-1. Retrieved 25 June 2011.
- ↑ Fluorescence Fundamentals. Invitrogen.com. Retrieved on 2011-06-25.
- ↑ Animation for the principle of fluorescence and UV-visible absorbance
- ↑ Fluorescent Probes. Piercenet.com. Retrieved on 2011-06-25.
- ↑ Lamture, JB; Wensel, TG (1995). "Intensely luminescent immunoreactive conjugates of proteins and dipicolinate-based polymeric Tb (III) chelates". Bioconjugate chemistry. 6 (1): 88–92. doi:10.1021/bc00031a010. PMID 7711110.
- ↑ Chalfie, M; Tu, Y; Euskirchen, G; Ward, WW; Prasher, DC (1994). "Green fluorescent protein as a marker for gene expression". Science. 263 (5148): 802–5. doi:10.1126/science.8303295. PMID 8303295.
- ↑ "The Micro Imager by Biospace Lab". Biospacelab.com. Archived from the original on 2009-01-05. Retrieved 2011-06-25.
- ↑ Evanko, Daniel (2005). "A 'flaky' but useful fluorophore". Nature Methods. 2 (3): 160–161. doi:10.1038/nmeth0305-160b.
- ↑ Zinchuk, Grossenbacher-Zinchuk (2009). "Recent advances in quantitative colocalization analysis: Focus on neuroscience". Prog Histochem Cytochem. 2 (3): 125–172. doi:10.1016/j.proghi.2009.03.001. PMID 19822255.
| Overview |
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Engineering |
| ||||||||||
| |||||||||||