Alkyne trimerisation
An alkyne trimerisation reaction is a 2+2+2 cyclization reaction in which three alkyne molecules react to form an aromatic compound. The reaction is 'pseudo' pericyclic since it has not been observed to occur without the assistance of metal catalysis; and the metal catalyst assembles the ring stepwise via intermediates which are not directly in between (in a geometric sense) the starting material and products.[1]
Introduction
History and scope
Berthelot reported the first example of cyclotrimerization leading to aromatic products in 1866, the cyclization of acetylene to benzene.[2] The reaction required very high temperatures to proceed (400 °C) and produced a complex mixture of products, and research in the field remain dormant until the late 1940s. In 1948, Reppe discovered that nickel could catalyze the formation of substituted benzenes from acetylenic compounds;[3] since his initial discovery, cyclotrimerizations to produce substituted benzenes have been catalyzed by no less than seventeen transition metals, including:
- Group 4 (Ti, Zr)[4]
- Group 5 (V, Nb)[5]
- Group 6 (Mo, W)[6]
- Group 8 (Fe, Ru, Os)[7]
- Group 9 (Co, Rh, Ir)[8]
- Group 10 (Pd)[9]
- Group 11 (Cu)[10]
Advantages of method
Control of the substitution pattern of the aromatic product is good in many cases, and cyclotrimerization can be used in cases when functionalization of pre-formed aromatic materials (through electrophilic aromatic substitution, for instance) is not selective. The reaction is highly chemoselective for triple bonds and can tolerate a wide variety of functional groups on the starting materials.
Disadvantages of method
For cotrimerizations involving two or three different acetylenes, a variety of regioisomers may form. Operationally, these reactions usually require elevated temperatures (>60 °C) and sometimes require irradiation to facilitate the dissociation of strongly binding carbon monoxide ligands. Catalyst deactivation can occur through the formation of stable, 18-electron η4-complexes incorporating cyclobutadiene,[11] cyclohexadiene, and arene ligands. The most problematic side products of the reaction are due to cyclotetramerization (leading to cyclooctatetraenes) and alkyne dimerization (leading to enynes).

Mechanism and stereochemistry
Prevailing view of mechanism
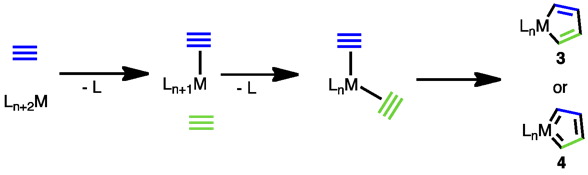
The most common mechanism for the cyclotrimerization of acetylenes begins with the formation of a metallocyclopentadiene complex. Oxidative cyclization of two coordinated alkyne units produces either metallocycle 3 or 4.[12] After dissociation of a third ligand and coordination of a third alkyne, two pathways are possible. Either alkyne insertion generates metallocycloheptatriene 5, or [4+2] cycloaddition generates bridged bicycle 6. The former pathway is questionable however, as reductive elimination from a metallocycloheptatriene to form an arene is symmetry forbidden.[13] A third pathway proposed for ruthenium catalysts involves formal [2+2] cycloaddition of the alkyne followed by rearrangement, reductive elimination, and arene decomplexation. The intermediacy of bicycle 7 was supported by DFT calculations.[14]
For cyclotrimerization reactions of asymmetrically substituted acetylenes, a number of regioisomeric products are possible. The substitution pattern about the product arene depends on two events: formation of the metallocyclopentadiene intermediate and incorporation of the third equivalent of alkyne. Although both head-to-head and tail-to-tail metallocyclopentadienes lead to 1, a number of acetylenic substrates selectively form regioisomers of type 2. Steric bulk on the alkyne coupling partners and catalyst have been invoked as the controlling elements of regioselectivity.[15]

Enantioselective variants
Chiral catalysts have been employed in combination with arynes to produce non-racemic amounts of atropisomeric products.[16] Selective cyclotrimerization of one of a pair of enantiotopic alkynes has also been facilitated by a chiral catalyst.[17]
Scope and limitations
Catalysts for cyclotrimerization are highly selective for triple bonds, which gives the reaction a fairly wide substrate scope; alcohols, ethers, amines, and carbonyl compounds (ketones, esters, amides, and carboxylic acids) are all well tolerated. Hoewever, the reaction is not generally compatible with unsaturated functionalities other than carbonyl compounds; nitriles, for instance, can react to form pyridines.[18]
In addition, some reactions are limited by catalyst deactivation via formation of stable, 18-electron η4-complexes.[19] Cyclobutadiene, cyclohexadiene, and arene complexes have all been observed as off-cycle, inactivate catalyst forms. In addition to high-order polymers and dimers and trimers, which originate from low regio- and chemoselectivities, enyne side products derived from alkyne dimerization have been observed. Rhodium catalysts are particularly adept at enyne formation (see below).[20] For nickel catalysis, formation of larger rings (particularly cyclooctatetraene) can be a problem.

Synthetic applications
Cyclization of diynes with a separate alkyne can provide fused ring systems with high atom economy. In a striking example in the synthesis of estrone, the diyne reactant shown below was combined with di(trimethylsilyl)acetylene to produce the benzocyclobutene product indicated.[21] Upon heating, ring-opening produced a quinone methide that participated in a subsequent intramolecular Diels-Alder reaction.
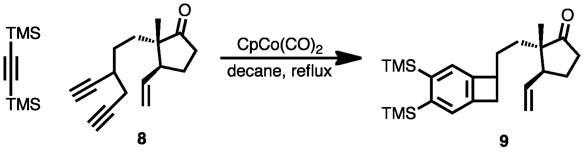
If the third alkyne unit is tethered to the first two, three rings can be created in a single step. In the example below in the synthesis of calomelanolactone, Wilkinson's catalyst was used to catalyze an intramolecular cotrimerization of the triyne shown.[22]

Crowded triynes can cyclize to products exhibiting helical chirality. In one example remarkable for the formation of three new aromatic rings in one step, the triyne shown is transformed into the helical product via treatment with cyclopentadienylcobalt dicarbonyl.[23] As of 2004, this process had yet to be rendered asymmetric, but the products could be separated through chiral HPLC.[23]

Comparison with other methods
Cyclotrimerization presents an alternative to the functionalization of pre-formed aromatic compounds through electrophilic or nucleophilic substitution, the regioselectivity of which can sometimes be difficult to control.
Other methods for the direct formation of aromatic rings from substituted, unsaturated precursors include the Dötz reaction, palladium-catalyzed [4+2] benzannulation of enynes with alkynes,[24] and Lewis-acid-mediated [4+2] cycloaddition of enynes with alkynes.[25] Cyclization of transient benzyne species with alkynes, catalyzed by palladium, can also produce substituted aromatic compounds.[26]

Experimental conditions
Although various experimental conditions have been used to perform this reaction, commercially available CpCo(CO)2 remains the most common catalyst used.[27] High temperatures and irradiation with light are often required in order to facilitate the dissociation of carbon monoxide from the catalyst.
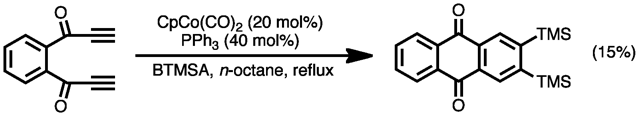
For instance, treatment of bis(trimethylsilyl)acetylene in the presence of CpCo(CO)2 in n-octane at reflux with 1,2-bis(propiolyl)benzene in diglyme added by slow addition under dry N2 atmosphere gave the anthraquinone shown, in 15% yield after removal of the solvent and unreacted volatiles, column chromatography on silica gel, and recrystallization.[28]
References
- ↑ Agenet, N.; Buisine, O.; Slowinski, F.; Gandon, V.; Aubert, C.; Malacria, M. (2007). "Cotrimerizations of Acetylenic Compounds". Org. React. 68: 1–302. doi:10.1002/0471264180.or068.01. ISBN 0471264180.
- ↑ Schetter, M. C. R. (1866). Hebd. Seances Acad. Sci. 62: 905.
- ↑ Reppe, W.; Schweckendiek, W. (1948). "Cyclisierende Polymerisation von Acetylen. III Benzol, Benzolderivate und hydroaromatische Verbindungen". Liebigs Ann. Chem. 560: 104. doi:10.1002/jlac.19485600104.
- ↑ Musso, F.; Solari, E.; Floriani, C. (1997). "Hydrocarbon Activation with Metal Halides: Zirconium Tetrachloride Catalyzing the Jacobsen Reaction and Assisting the Trimerization of Alkynes via the Formation of η6-Arene−Zirconium(IV) Complexes". Organometallics. 16 (22): 4889. doi:10.1021/om970438g.
- ↑ Rodriguez, J. G.; Martin-Villamil, R.; Fonseca, I. (1997). J. Chem. Soc., Perkin Trans. 1: 945.
- ↑ Sakurai, H.; Nakadaira, Y.; Hosomi, A.; Eriyama, Y.; Hirama, K.; Kabuto, C. (1984). "Chemistry of organosilicon compounds. 193. Intramolecular cyclotrimerization of macrocylic and acyclic triynes with Group 6 metal carbonyls. The formation of fulvene and benzene". J. Am. Chem. Soc. 106 (26): 8315. doi:10.1021/ja00338a063.
- ↑ Reppe, W.; Vetter, H. (1953). Liebigs Ann. Chem. 585: 133.
- ↑ Amer, I.; Bernstein, T.; Eisen, M.; Blum, J.; Vollhardt, K. P. C. (1990). "Oligomerization of alkynes by the RhCl3-aliquat 336 catalyst system Part 1. Formation of benzene derivatives". J. Mol. Catal. 60 (3): 313. doi:10.1016/0304-5102(90)85254-F.
- ↑ Lee, C. L.; Hunt, C. T.; Balch, A. L. (1981). "Novel reactions of metal-metal bonds. Reactions of Pd2{(C6H5)2PCH2P(C6H5)2}2Cl2 with acetylenes, olefins, and isothiocyanates". Inorg. Chem. 20 (8): 2498. doi:10.1021/ic50222a026.
- ↑ Aalten, H. L.; van Koten, G.; Riethorst, E.; Stam, C. H. (1989). "The Hurtley reaction. 2. Novel complexes of disubstituted acetylenes with copper(I) benzoates having a reactive ortho carbon-chlorine or carbon-bromine bond. X-ray structural characterization of tetrakis(2-chlorobenzoato)bis(diethyl acetylenedicarboxylate)tetracopper(I)". Inorg. Chem. 28 (22): 4140. doi:10.1021/ic00321a020.
- ↑ Yamazaki, H.; Hagihara, N. (1970). "Preparations and reactions of alkylcobalt complexes and diphenylacetylenecobalt complexes having a π-cyclopentadienyl group as a ligand". J. Organomet. Chem. 21 (2): 431. doi:10.1016/S0022-328X(00)83645-1.
- ↑ Collman, J. P.; Kang, J. W.; Little, W. F.; Sullivan, M. F. (1968). "Metallocyclopentadiene complexes of iridium and rhodium and their role in the catalytic cyclotrimerization of disubstituted acetylenes". Inorg. Chem. 7 (7): 1298. doi:10.1021/ic50065a007.
- ↑ Hardesty, J. H.; Koerner, J. B.; Albright, T. A.; Lee, G. B. (1999). "Theoretical Study of the Acetylene Trimerization with CpCo". J. Am. Chem. Soc. 121 (25): 6055. doi:10.1021/ja983098e.
- ↑ Kirchner, K.; Calhorda, M. J.; Schmid, R.; Veiros, L. F. (2003). "Mechanism for the cyclotrimerization of alkynes and related reactions catalyzed by CpRuCl". J. Am. Chem. Soc. 125 (38): 11721–9. doi:10.1021/ja035137e. PMID 13129377.
- ↑ Ozerov, O. V.; Patrick, B. O.; Ladipo, F. T. (2000). "Highly Regioselective [2 + 2 + 2] Cycloaddition of Terminal Alkynes Catalyzed by η6-Arene Complexes of Titanium Supported by Dimethylsilyl-Bridgedp-tert-Butyl Calix[4]arene Ligand". J. Am. Chem. Soc. 122 (27): 6423. doi:10.1021/ja994543o.
- ↑ Gutnov, A.; Heller, B.; Fisher, C.; Drexler, H. J.; Spannenberg, A.; Sundermann, B.; Sundermann, C. (2004). Angew. Chem. Int. Ed. Engl. 43: 2.
- ↑ Sato, Y.; Nishimata, T.; Mori, M. (1997). "Novel Synthesis of Heterocycles Using Nickel(0)-catalyzed [2+2+2] Cocyclization: Catalytic Asymmetric Synthesis of Isoindoline and Isoquinoline Derivatives". Heterocycles. 44: 443. doi:10.3987/COM-96-S43.
- ↑ Varela, Jesus; Saa, Carlos (March 20, 2003). "Construction of Pyridine Rings by Metal-Mediated [2 + 2 + 2] Cycloaddition†". Chemistry Review. doi:10.1021/cr030677f. Retrieved April 11, 2015.
- ↑ Kölle, U.; Fuss, B. (1986). "Pentamethylcyclopentadienyl-Übergangsmetall-Komplexe, X. Neue Co-Komplexe aus η5-C5Me5Co-Fragmenten und Acetylenen". Chem. Ber. 119: 116. doi:10.1002/cber.19861190112.
- ↑ Ardizzoia, G. A.; Brenna, S.; Cenini, S.; LaMonica, G.; Masciocchi, N.; Maspero, A. (2003). "Oligomerization and Polymerization of Alkynes Catalyzed by Rhodium(I) Pyrazolate Complexes". J. Mol. Catal. A: Chemical. 204–205: 333–340. doi:10.1016/S1381-1169(03)00315-7.
- ↑ Funk, R. L.; Vollhardt, K. P. C. (1980). "Transition-metal-catalyzed alkyne cyclizations. A cobalt-mediated total synthesis of dl-estrone". J. Am. Chem. Soc. 102 (16): 5253–5261. doi:10.1021/ja00536a023.
- ↑ Neeson, S. J.; Stevenson, P. J. (1988). "Rhodium catalysed [2+2+2] cycloadditions- an efficient regiospecific route to calomelanolactone". Tetrahedron Lett. 29 (7): 813. doi:10.1016/S0040-4039(00)80217-8.
- 1 2 Teply, F.; Stara, I. G.; Stary, I.; Kollarovic, A.; Saman, D.; Rulisek, L.; Fiedler, P. (2002). "Synthesis of 5-, 6-, and 7helicene via Ni(0)- or Co(I)-catalyzed isomerization of aromatic cis,cis-dienetriynes". J. Am. Chem. Soc. 124 (31): 9175–80. doi:10.1021/ja0259584. PMID 12149022.
- ↑ Gevorgyan, V.; Takeda, A.; Homma, M.; Sadayori, N.; Radhakrishnan, U.; Yamamoto, Y. (1999). "Palladium-Catalyzed [4+2]Cross-Benzannulation Reaction of Conjugated Enynes with Diynes and Triynes". J. Am. Chem. Soc. 121 (27): 6391. doi:10.1021/ja990749d.
- ↑ Wills, M. S. B.; Danheiser, R. L. (1998). "Intramolecular [4 + 2] Cycloaddition Reactions of Conjugated Ynones. Formation of Polycyclic Furans via the Generation and Rearrangement of Strained Heterocyclic Allenes". J. Am. Chem. Soc. 120 (36): 9378. doi:10.1021/ja9819209.
- ↑ Sato, Y.; Tamura, T.; Mori, M. (2004). "Arylnaphthalene lignans through Pd-Catalyzed 2+2+2 cocyclization of arynes and diynes: total synthesis of Taiwanins C and E". Angew. Chem. Int. Ed. Engl. 43 (18): 2436–40. doi:10.1002/anie.200453809. PMID 15114584.
- ↑ Dominguez, Gema; Perez-Castells, Javier (March 23, 2011). "Recent advances in [2+2+2] cycloaddition reactions". Royal Society of Chemistry. doi:10.1039/C1CS15029D. Retrieved April 13, 2015.
- ↑ Hillard III, R. L.; Vollhardt, K. P. C. (1977). "Substituted benzocyclobutenes, indans, and tetralins via cobalt-catalyzed cooligomerization of .alpha.,.omega.-diynes with substituted acetylenes. Formation and synthetic utility of trimethylsilylated benzocycloalkenes". J. Am. Chem. Soc. 99 (12): 4058. doi:10.1021/ja00454a026.