Chemical shift index
The chemical shift index or CSI is a widely employed technique in protein nuclear magnetic resonance spectroscopy that can be used to display and identify the location (i.e. start and end) as well as the type of protein secondary structure (beta strands, helices and random coil regions) found in proteins using only backbone chemical shift data [1][2] The technique was invented by Dr. David Wishart in 1992 for analyzing 1Hα chemical shifts and then later extended by him in 1994 to incorporate 13C backbone shifts. The original CSI method makes use of the fact that 1Hα chemical shifts of amino acid residues in helices tends to be shifted upfield (i.e. towards the right side of a NMR spectrum) relative to their random coil values and downfield (i.e. towards the left side of a NMR spectrum) in beta strands. Similar kinds of upfield/downfiled trends are also detectable in backbone 13C chemical shifts.
Implementation
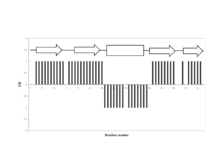
The CSI is a graph-based technique that essentially employs an amino acid-specific digital filter to convert every assigned backbone chemical shift value into a simple three-state (-1, 0, +1) index. This approach generates a more easily understood and much more visually pleasing graph of protein chemical shift values. In particular, if the upfield 1Hα chemical shift (relative to an amino acid-specific random coil value) of a certain residue is > 0.1 ppm, then that amino acid residue is assigned a value of -1. Similarly, if the downfield 1Hα chemical shift of a certain amino acid residue is > 0.1 ppm then that residue is assigned a value of +1. If an amino acid residue’s chemical shift is not shifted downfield or upfield by a sufficient amount (i.e. <0.1 ppm), it is given a value of 0. When this 3-state index is plotted as a bar graph over the full length of the protein sequence, simple inspection can allow one to identify beta strands (clusters of +1 values), alpha helices (clusters of -1 values), and random coil segments (clusters of 0 values). A list of the amino acid-specific random coil chemical shifts for CSI calculations is given in Table 1. An example of a CSI graph for a small protein is shown in Figure 1 with the arrows located above the black bars indicating locations of the beta strands and the rectangular box indicating the location of a helix.
| Amino Acid | 1Hα random coil shift (ppm) | Amino Acid | 1Hα RC shift random coil shift (ppm) |
|---|---|---|---|
| Ala (A) | 4.35 | Met (M) | 4.52 |
| Cys (C) | 4.65 | Asn (N) | 4.75 |
| Asp (D) | 4.76 | Pro (P) | 4.44 |
| Glu (E) | 4.29 | Gln (Q) | 4.37 |
| Phe (F) | 4.66 | Arg (R) | 4.38 |
| Gly (G) | 3.97 | Ser (S) | 4.50 |
| His (H) | 4.63 | Thr (T) | 4.35 |
| Ile (I) | 3.95 | Val (V) | 3.95 |
| Lys (K) | 4.36 | Trp (W) | 4.70 |
| Leu (L) | 4.17 | Tyr (Y) | 4.60 |
Performance
Using only 1Hα chemical shifts and simple clustering rules (clusters of 3 or more vertical bars for beta strands and clusters of 4 or more vertical bars for alpha helices), the CSI is typically 75-80% accurate in the identification of secondary structures.[2][3][4][5] This performance depends partly on the quality of the NMR data set as well as the technique (manual or programmatic) used to identify the protein secondary structures. As noted above, a consensus CSI method that filters upfield/downfield chemical shift changes in 13Cα, 13Cβ, and 13C' atoms in a similar manner to 1Hα shifts has also been developed.[2] The consensus CSI combines the CSI plots from backbone 1H and 13C chemical shifts to generate a single CSI plot. It can be up to 85-90% accurate.[5]
History
The link between protein chemical shifts and protein secondary structure (specifically alpha helices) was first described by John Markley and colleagues in 1967.[6] With the development of modern 2-dimensional NMR techniques, it became possible to measure more protein chemical shifts. With more peptides and proteins were being assigned in the early 1980s it soon became obvious that amino acid chemical shifts were sensitive not only to helical conformations, but also to β-strand conformations. Specifically, the secondary 1Hα chemical shifts of all amino acids exhibit a clear upfield trend on helix formation and an obvious downfield trend on β-sheet formation.[7][8] By the early 1990s, a sufficient body of 13C and 15N chemical shift assignments for peptides and proteins had been collected to determine that similar upfield/downfield trends were evident for essentially all backbone 13Cα, 13Cβ, 13C', 1HN and 15N (weakly) chemical shifts.[9][10] It was these rather striking chemical shift trends that were exploited in the development of the chemical shift index.
Limitations
The CSI method is not without some shortcomings. In particular, its performance drops if chemical shift assignments are mis-referenced or incomplete. It is also quite sensitive to the choice of random coil shifts used to calculate the secondary shifts[5] and it generally identifies alpha helices (>85% accuracy) better than beta strands (<75% accuracy) regardless of the choice of random coil shifts.[5] Furthermore, the CSI method does not identify other kinds of secondary structures, such as β-turns. Because of these shortcomings, a number of alternative CSI-like approaches have been proposed. These include: 1) a prediction method that employs statistically derived chemical shift/structure potentials (PECAN);[11] 2) a probabilistic approach to secondary structure identification (PSSI);[12] 3) a method that combines secondary structure predictions from sequence data and chemical shift data (PsiCSI),[13] 4) a secondary structure identification approach that uses pre-specified chemical shift patterns (PLATON)[14] and 5) a two-dimensional cluster analysis method known as 2DCSi.[15] The performance of these newer methods is generally slightly better (2-4%) than the original CSI method.
Utility
Since its original description in 1992, the CSI method has been used to characterize the secondary structure of thousands of peptides and proteins. Its popularity is largely due to the fact that it is easy to understand and can be implemented without the need for specialized computer programs. Even though the CSI method can be easily performed manually, a number of commonly used NMR data processing programs such as NMRView,[16] NMR structure generation web servers such as CS23D[17] as well as various NMR data analysis web servers such as RCI,[18] Preditor[19] and PANAV [20] have incorporated the CSI method into their software.
See also
- Chemical Shift
- Random Coil Index
- Protein NMR
- Protein Chemical Shift Re-Referencing
- Protein secondary structure
- Protein Chemical Shift Prediction
- NMR
- Nuclear magnetic resonance spectroscopy
- Protein nuclear magnetic resonance spectroscopy
- Protein
References
- ↑ Wishart DS, Sykes BD, Richards FM (February 1992). "The chemical shift index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy". Biochemistry. 31 (6): 1647–51. doi:10.1021/bi00121a010. PMID 1737021.
- 1 2 3 Wishart, David S.; Sykes, Brian D. (1994). "The 13C Chemical-Shift Index: A simple method for the identification of protein secondary structure using 13C chemical-shift data". Journal of Biomolecular NMR. 4 (2): 171–80. doi:10.1007/BF00175245. PMID 8019132.
- ↑ Wishart DS, Case DA (2001). "Use of chemical shifts in macromolecular structure determination". Methods in Enzymology. 338: 3–34. doi:10.1016/s0076-6879(02)38214-4. PMID 11460554.
- ↑ Mielke SP, Krishnan VV (April 2009). "Characterization of protein secondary structure from NMR chemical shifts". Progress in Nuclear Magnetic Resonance Spectroscopy. 54 (3–4): 141–165. doi:10.1016/j.pnmrs.2008.06.002. PMC 2766081
 . PMID 20160946.
. PMID 20160946. - 1 2 3 4 Wishart DS (February 2011). "Interpreting protein chemical shift data". Progress in Nuclear Magnetic Resonance Spectroscopy. 58 (1–2): 62–87. doi:10.1016/j.pnmrs.2010.07.004. PMID 21241884.
- ↑ Markley JL, Meadows DH, Jardetzky O (July 1967). "Nuclear magnetic resonance studies of helix-coil transitions in polyamino acids". Journal of Molecular Biology. 27 (1): 25–40. doi:10.1016/0022-2836(67)90349-X. PMID 6033611.
- ↑ Clayden, N.J; Williams, R.J.P (1982). "Peptide group shifts". Journal of Magnetic Resonance. 49 (3): 383. Bibcode:1982JMagR..49..383C. doi:10.1016/0022-2364(82)90252-9.
- ↑ Pardi A, Wagner G, Wüthrich K (December 1983). "Protein conformation and proton nuclear-magnetic-resonance chemical shifts". European Journal of Biochemistry. 137 (3): 445–54. doi:10.1111/j.1432-1033.1983.tb07848.x. PMID 6198174.
- ↑ Wishart DS, Sykes BD, Richards FM (November 1991). "Relationship between nuclear magnetic resonance chemical shift and protein secondary structure". Journal of Molecular Biology. 222 (2): 311–33. doi:10.1016/0022-2836(91)90214-Q. PMID 1960729.
- ↑ Spera, Silvia; Bax, Ad (1991). "Empirical correlation between protein backbone conformation and Cα and Cβ 13C nuclear magnetic resonance chemical shifts". Journal of the American Chemical Society. 113 (14): 5490–2. doi:10.1021/ja00014a071. INIST:5389018.
- ↑ Eghbalnia HR, Wang L, Bahrami A, Assadi A, Markley JL (May 2005). "Protein energetic conformational analysis from NMR chemical shifts (PECAN) and its use in determining secondary structural elements". Journal of Biomolecular NMR. 32 (1): 71–81. doi:10.1007/s10858-005-5705-1. PMID 16041485.
- ↑ Wang Y, Jardetzky O (April 2002). "Probability-based protein secondary structure identification using combined NMR chemical-shift data". Protein Science. 11 (4): 852–61. doi:10.1110/ps.3180102. PMC 2373532. PMID 11910028.
- ↑ Hung LH, Samudrala R (February 2003). "Accurate and automated classification of protein secondary structure with PsiCSI". Protein Science. 12 (2): 288–95. doi:10.1110/ps.0222303. PMC 2312422. PMID 12538892.
- ↑ Labudde D, Leitner D, Krüger M, Oschkinat H (January 2003). "Prediction algorithm for amino acid types with their secondary structure in proteins (PLATON) using chemical shifts". Journal of Biomolecular NMR. 25 (1): 41–53. PMID 12566998.
- ↑ Wang CC, Chen JH, Lai WC, Chuang WJ (May 2007). "2DCSi: identification of protein secondary structure and redox state using 2D cluster analysis of NMR chemical shifts". Journal of Biomolecular NMR. 38 (1): 57–63. doi:10.1007/s10858-007-9146-x. PMID 17333485.
- ↑ Johnson BA, Blevins RA (September 1994). "NMR View: A computer program for the visualization and analysis of NMR data". Journal of Biomolecular NMR. 4 (5): 603–14. doi:10.1007/BF00404272. PMID 22911360.
- ↑ Wishart DS, Arndt D, Berjanskii M, Tang P, Zhou J, Lin G (July 2008). "CS23D: a web server for rapid protein structure generation using NMR chemical shifts and sequence data". Nucleic Acids Research. 36 (Web Server issue): W496–502. doi:10.1093/nar/gkn305. PMC 2447725. PMID 18515350.
- ↑ Berjanskii MV, Wishart DS (July 2007). "The RCI server: rapid and accurate calculation of protein flexibility using chemical shifts". Nucleic Acids Research. 35 (Web Server issue): W531–7. doi:10.1093/nar/gkm328. PMC 1933179. PMID 17485469.
- ↑ Berjanskii MV, Neal S, Wishart DS (July 2006). "PREDITOR: a web server for predicting protein torsion angle restraints". Nucleic Acids Research. 34 (Web Server issue): W63–9. doi:10.1093/nar/gkl341. PMC 1538894. PMID 16845087.
- ↑ Wang B, Wang Y, Wishart DS (June 2010). "A probabilistic approach for validating protein NMR chemical shift assignments". Journal of Biomolecular NMR. 47 (2): 85–99. doi:10.1007/s10858-010-9407-y. PMID 20446018.
External links
- CSI calculations via RCI webserver http://randomcoilindex.com
- CSI calculations via Preditor webserver http://preditor.ca
- Stand-alone CSI program for Linux/Unix http://www.bionmr.ualberta.ca/sykes/software/csi/latest/csi.html
- Chemical shift rereferencing for CSI calculations by Shiftcor http://shiftcor.wishartlab.com/
- Chemical shift rereferencing for CSI calculations by PANAV https://web.archive.org/web/20140413150003/http://www.wishartlab.com/web_servers/panav