Amyloid beta
| Amyloid beta peptide (beta-APP) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
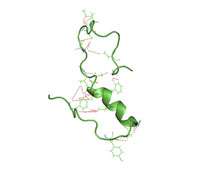 A partially folded structure of amyloid beta(1 40) in an aqueous environment (pdb 2lfm)[1] | |||||||||
| Identifiers | |||||||||
| Symbol | APP | ||||||||
| Pfam | PF03494 | ||||||||
| InterPro | IPR013803 | ||||||||
| SCOP | 2lfm | ||||||||
| SUPERFAMILY | 2lfm | ||||||||
| TCDB | 1.C.50 | ||||||||
| OPM superfamily | 369 | ||||||||
| OPM protein | 2y3k | ||||||||
| |||||||||
| amyloid beta (A4) precursor protein (peptidase nexin-II, Alzheimer disease) | |
|---|---|
|
Processing of the amyloid precursor protein | |
| Identifiers | |
| Symbol | APP |
| Alt. symbols | AD1 |
| Entrez | 351 |
| HUGO | 620 |
| OMIM | 104760 |
| RefSeq | NM_000484 |
| UniProt | P05067 |
| Other data | |
| Locus | Chr. 21 q21.2 |

Amyloid beta (Aβ or Abeta) denotes peptides of 36–43 amino acids that are crucially involved in Alzheimer's disease as the main component of the amyloid plaques found in the brains of Alzheimer patients. The peptides result from the amyloid precursor protein (APP), which is cleaved by beta secretase and gamma secretase to yield Aβ. Aβ molecules can aggregate to form flexible soluble oligomers which may exist in several forms. It is now believed that certain misfolded oligomers (known as "seeds") can induce other Aβ molecules to also take the misfolded oligomeric form, leading to a chain reaction akin to a prion infection. The seeds or the resulting amyloid plaques are toxic to nerve cells. The other protein implicated in Alzheimer's disease, tau protein, also forms such prion-like misfolded oligomers, and there is some evidence that misfolded Aβ can induce tau to misfold.[2][3]
A recent study suggested that APP and its amyloid potential is of ancient origins, dating as far back as early deuterostomes.[4]
Normal function
The normal function of Aβ is not well understood.[5] Though some animal studies have shown that the absence of Aβ does not lead to any loss of physiological function,[6][7] several potential activities have been discovered for Aβ, including activation of kinase enzymes,[8][9] protection against oxidative stress,[10][11] regulation of cholesterol transport,[12][13] functioning as a transcription factor,[14][15] and anti-microbial activity (potentially associated with Aβ's pro-inflammatory activity).[16]
Per a study published in the 2016 May 25 issue of Science Translational Medicine, Amyloid-beta protein may fight other diseases, attacking pathogens in mice and worms.[17][18]
The glymphatic system clears metabolic waste from the mammalian brain, and in particular beta amyloids.[19] The rate of removal is significantly increased during sleep.[20] However the significance of the glymphatic system is unknown in clearance of Aβ.[21]
Disease associations
Aβ is the main component of amyloid plaques (extracellular deposits found in the brains of patients with Alzheimer's disease).[22] Similar plaques appear in some variants of Lewy body dementia and in inclusion body myositis (a muscle disease), while Aβ can also form the aggregates that coat cerebral blood vessels in cerebral amyloid angiopathy. The plaques are composed of a tangle of regularly ordered fibrillar aggregates called amyloid fibers,[23] a protein fold shared by other peptides such as the prions associated with protein misfolding diseases.
Alzheimer's disease
Recent research suggests that soluble oligomeric forms of the peptide may be causative agents in the development of Alzheimer's disease.[24][25] It is generally believed that Aβ oligomers are the most toxic.[26] The ion channel hypothesis postulates that oligomers of soluble, non-fibrillar Aβ form membrane ion channels allowing the unregulated calcium influx into neurons[27] that underlies disrupted calcium ion homeostasis and apoptosis seen in Alzheimer's disease.[28][29] A number of genetic, cell biology, biochemical and animal studies support the concept that Aβ plays a central role in the development of Alzheimer’s disease pathology.[30][31]
Brain Aβ is elevated in patients with sporadic Alzheimer’s disease. Aβ is the main constituent of brain parenchymal and vascular amyloid; it contributes to cerebrovascular lesions and is neurotoxic.[30][31][32][33] It is unresolved how Aβ accumulates in the central nervous system and subsequently initiates the disease of cells. Some researchers have found that the Aβ oligomers induce some of the symptoms of Alzheimer's Disease by competing with insulin for binding sites on the insulin receptor, thus impairing glucose metabolism in the brain.[34] Significant efforts have been focused on the mechanisms responsible for Aβ production, including the proteolytic enzymes gamma- and β-secretases which generate Aβ from its precursor protein, APP (amyloid precursor protein).[35][36][37][38] Aβ circulates in plasma, cerebrospinal fluid (CSF) and brain interstitial fluid (ISF) mainly as soluble Aβ40[30][39] Senile plaques contain both Aβ40 and Aβ42,[40] while vascular amyloid is predominantly the shorter Aβ40. Several sequences of Aβ were found in both lesions.[41][42][43] Generation of Aβ in the CNS may take place in the neuronal axonal membranes after APP-mediated axonal transport of β-secretase and presenilin-1.[44]
Increases in either total Aβ levels or the relative concentration of both Aβ40 and Aβ42 (where the former is more concentrated in cerebrovascular plaques and the latter in neuritic plaques)[45] have been implicated in the pathogenesis of both familial and sporadic Alzheimer's disease. Due to its more hydrophobic nature, the Aβ42 is the most amyloidogenic form of the peptide. However the central sequence KLVFFAE is known to form amyloid on its own, and probably forms the core of the fibril.
The "amyloid hypothesis", that the plaques are responsible for the pathology of Alzheimer's disease, is accepted by the majority of researchers but is by no means conclusively established. An alternative hypothesis is that amyloid oligomers rather than plaques are responsible for the disease.[26][46] Mice that are genetically engineered to express oligomers but not plaques (APPE693Q) develop the disease. Furthermore, mice that are in addition engineered to convert oligomers into plaques (APPE693Q X PS1ΔE9), are no more impaired than the oligomer only mice.[47] Intra-cellular deposits of tau protein are also seen in the disease, and may also be implicated, as has aggregation of alpha synuclein.
Currently, research is being done using biomarkers and ELISA tests to determine levels of amyloid beta so blood tests can detect Alzheimer's Disease in its early stages. Of 24 biomarkers, 3 were confirmed to be reliable identification markers of AD patients.[48]
Formation
Aβ is formed after sequential cleavage of the amyloid precursor protein (APP), a transmembrane glycoprotein of undetermined function. APP can be cleaved by the proteolytic enzymes α-, β- and γ-secretase; Aβ protein is generated by successive action of the β and γ secretases. The γ secretase, which produces the C-terminal end of the Aβ peptide, cleaves within the transmembrane region of APP and can generate a number of isoforms of 30-51 amino acid residues in length.[49] The most common isoforms are Aβ40 and Aβ42; the longer form is typically produced by cleavage that occurs in the endoplasmic reticulum, while the shorter form is produced by cleavage in the trans-Golgi network.[50] The Aβ40 form is the more common of the two, but Aβ42 is the more fibrillogenic and is thus associated with disease states. Mutations in APP associated with early-onset Alzheimer's have been noted to increase the relative production of Aβ42, and thus one suggested avenue of Alzheimer's therapy involves modulating the activity of β and γ secretases to produce mainly Aβ40.[51] Aβ is destroyed by several amyloid-degrading enzymes including neprilysin.[52]
Genetics
Autosomal-dominant mutations in APP cause hereditary early-onset Alzheimer's disease (a.k.s. familial AD). This form of AD accounts for no more than 10% of all cases, and the vast majority of AD is not accompanied by such mutations.[53] However, familial Alzheimer disease is likely to result from altered proteolytic processing.
The gene for amyloid precursor protein is located on chromosome 21, and accordingly people with Down syndrome have a very high incidence of Alzheimer's disease.
Structure and toxicity
Amyloid beta is commonly thought to be intrinsically unstructured, meaning that in solution it does not acquire a unique tertiary fold but rather populates a set of structures. As such, it cannot be crystallized and most structural knowledge on amyloid beta comes from NMR and molecular dynamics. Early NMR-derived models of a 26-aminoacid polypeptide from amyloid beta (Aβ 10-35) show a collapsed coil structure devoid of significant secondary structure content.[54] However, the most recent (2012) NMR structure of (Aβ 1-40) has significant secondary and tertiary structure.[1] Replica exchange molecular dynamics studies suggested that amyloid beta can indeed populate multiple discrete structural states;[55] more recent studies identified a multiplicity of discrete conformational clusters by statistical analysis.[56] By NMR-guided simulations, amyloid beta 1-40 and amyloid beta 1-42 also seem to feature highly different conformational states,[57] with the C-terminus of amyloid beta 1-42 being more structured than that of the 1-40 fragment.
Structural information on the oligomeric state of amyloid beta is still sparse as of 2010. Low-temperature and low-salt conditions allowed to isolate pentameric disc-shaped oligomers devoid of beta structure.[58] In contrast, soluble oligomers prepared in the presence of detergents seem to feature substantial beta sheet content with mixed parallel and antiparallel character, different from fibrils;[59] computational studies suggest an antiparallel beta-turn-beta motif instead for membrane-embedded oligomers.[60]
The mechanism by which amyloid beta may damage and kill neurons is by generating reactive oxygen species during the process of its self-aggregation. When this occurs on the membrane of neurons it causes lipid peroxidation and the generation of a toxic aldehyde called 4-hydroxynonenal which, in turn, impairs the function of ion-motive ATPases, glucose transporters and glutamate transporters. As a result, amyloid beta promotes depolarization of the synaptic membrane, excessive calcium influx and mitochondrial impairment.[61]
Intervention strategies
Researchers in Alzheimer's disease have identified five strategies as possible interventions against amyloid:[62]
- β-Secretase inhibitors. These work to block the first cleavage of APP inside of the cell, at the endoplasmic reticulum.
- γ-Secretase inhibitors (e. g. semagacestat). These work to block the second cleavage of APP in the cell membrane and would then stop the subsequent formation of Aβ and its toxic fragments.
- Selective Aβ42 lowering agents (e. g. tarenflurbil). These modulate γ-secretase to reduce Aβ42 production in favor of other (shorter) Aβ versions.
β- and γ-secretase are responsible for the generation of Aβ from the release of the intracellular domain of APP, meaning that compounds that can partially inhibit the activity of either β- and γ-secretase are highly sought after. In order to initiate partial inhibition of β- and γ-secretase, a compound is needed that can block the large active site of aspartyl proteases while still being capable of bypassing the blood-brain barrier. To date, human testing has been avoided due to concern that it might interfere with signaling via Notch proteins and other cell surface receptors.
- Immunotherapy. This stimulates the host immune system to recognize and attack Aβ, or provide antibodies that either prevent plaque deposition or enhance clearance of plaques or Aβ oligomers. Oligomerization is a chemical process that converts individual molecules into a chain consisting of a finite number of molecules. Prevention of oligomerization of Aβ has been exemplified by active or passive Aβ immunization. In this process antibodies to Aβ are used to decrease cerebral plaque levels. This is accomplished by promoting microglial clearance and/or redistributing the peptide from the brain to systemic circulation. One such beta-amyloid vaccine that is currently in clinical trials is CAD106.[63] Immunization with synthetic Aβ1-42 has been shown to be beneficial in mice and displays low toxicity; however human trials have shown no significant differences. Thus, it is not yet effective in humans and requires further research. Specific findings show that the 20 amino acid SDPM1 protein binds tetramer forms of Aβ(1-40)- and Aβ(1-42)-amyloids and blocks subsequent Aβ amyloid aggregation. It is important to note that this study was done in mice and that while it prevents further development of neuropathology it did not result in an improvement in cognitive performance. Lastly, Aβ42 immunization resulted in the clearance of amyloid plaques in patients with Alzheimer's disease but did not prevent progressive neurodegeneration.[64] A more recent study showed FDA-approved cancer drugs, PD-1 inhibitors, may be effective at clearing Aβ plaques and improving cognitive performance in a mouse model of Alzheimer's disease.[65]
- Anti-aggregation agents[66] such as apomorphine. These prevent Aβ fragments from aggregating or clear aggregates once they are formed.[67] Studies comparing synthetic to recombinant Aβ42 in assays measuring rate of fibrillation, fibril homogeneity, and cellular toxicity showed that recombinant Aβ42 had a faster fibrillation rate and greater toxicity than synthetic Amyloid beta 1-42 peptide.[68] This observation combined with the irreproducibility of certain Aβ42 experimental studies has been suggested to be responsible for the lack of progress in Alzheimer’s research.[69] Consequently, there has been renewed efforts to manufacture Aβ42 and other amyloid peptides at unprecedented (>99%) purity[70]
There is some indication that supplementation of the hormone melatonin may be effective against amyloid. Melatonin interacts with amyloid beta and inhibits its aggregation[71][72][73] This anti-aggregatory activity occurs only through an interaction with dimers of the soluble amyloid beta peptide. Melatonin does not reverse fibril formation or oligomers of amyloid beta once they are formed. This is supported by experiments in transgenic mice which suggest that melatonin has the potential to prevent amyloid deposition if administered early in life, but it may not be efficacious to revert amyloid deposition or treat Alzheimer's disease.
This connection with melatonin, which regulates sleep, is strengthened by the recent research showing that the wakefulness inducing hormone orexin influences amyloid beta (see below).[74] Interestingly, animal experiments show that melatonin may also correct mild elevations of cholesterol which is also an early risk factor for amyloid formation in human brain.[75]
The cannabinoid HU-210 has been shown to prevent amyloid beta-promoted inflammation.[76] The endocannabinoids anandamide and noladin ether have also been shown to be neuroprotective against amyloid beta in vitro.[77]
It has been shown that high-cholesterol diets tend to increase Aβ pathology in animals.[78] Modulating cholesterol homeostasis has yielded results that show that chronic use of cholesterol-lowering drugs, such as the statins, is associated with a lower incidence of AD. In APP genetically modified mice, cholesterol-lowering drugs have been shown to reduce overall pathology. While the mechanism is poorly understood it appears that cholesterol-lowering drugs have a direct effect on APP processing.[79][80]
Chelation therapy, which involves the removal of heavy metals from the body, has also been shown to be beneficial in lowering amyloid plaque levels. This is because Aβ aggregation is somewhat dependent on the metal ions copper and zinc. Zinc in synaptic vesicles, which is under the control of the zinc transporter ZnT3, plays a major role in Aβ formation. The expression of the ZnT3 is significantly lower in Alzheimer’s patients compared to healthy patients. Mice without ZnT3 were found to have much higher plaque formation. Further promoting this concept, Aβ deposition was impeded in APP transgenic mice treated with the antibiotic clioquinol, a known copper/zinc chelator.[80]
Drug therapy has been another approach to treatment. Memantine is an Alzheimer’s drug which has received widespread approval. It is a non-competitive N-methyl-D-aspartate (NMDA) channel blocker. By binding to the NMDA receptor with a higher affinity than Mg2+ ions, memantine is able to inhibit the prolonged influx of Ca2+ ions, particularly from extrasynaptic receptors, which forms the basis of neuronal excitotoxicity. It is an option for the management of patients with moderate to severe Alzheimer's Disease (modest effect). The study showed that 20 mg/day improved cognition, functional ability and behavioural symptoms in patient population.[81]
Another drug under research is liraglutide, which is typically used as a diabetes drug. Treatment with liraglutide yielded cognitive benefits that included improved object and spatial recognition. It enhanced induction and maintenance of long term potentiation (LTP) and paired-pulse facilitation (PPF) in both APP/PS1 and non-genetically altered mice. Other histological benefits include a reduced inflammatory response and an increase in the number of young neurons in the dentate gyrus. The β-amyloid level was also found to be significantly reduced.[67]
Circadian rhythm of amyloid beta
A 2009 report demonstrated that amyloid beta production follows a circadian rhythm, rising when an animal (mouse) or person is awake and falling during sleep.[74] The wakefulness-promoting neuroprotein orexin was shown to be necessary for the circadian rhythm of amyloid beta production.[74] The report suggested that excessive periods of wakefulness (i.e. due to sleep debt) could cause chronic build-up of amyloid beta, which could hypothetically lead to Alzheimer's disease.[74] This is consistent with recent findings that chronic sleep deprivation is associated with early onset Alzheimer's disease.[74]
Melatonin is also involved in circadian rhythm maintenance. Notably, melatonin has been connected with the "sundowning" phenomenon, in which Alzheimer's disease patients that have amyloid plaques in the hypothalamus exhibit exacerbation of Alzheimer's disease symptoms late in the day.[82] This "sundowning" phenomenon could be directly or indirectly related to the recently discovered continuous increase in amyloid beta throughout the day.
Measuring amyloid beta
Imaging compounds, notably Pittsburgh compound B, (6-OH-BTA-1, a thioflavin), can selectively bind to amyloid beta in vitro and in vivo. This technique, combined with PET imaging, has been used to image areas of plaque deposits in Alzheimer's patients.
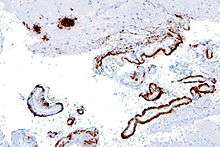
Post mortum or in tissue biopsies
There are many different ways to measure Amyloid beta. It can be measured semi-quantitatively with immunostaining, which also allows one to determine location. Amyloid beta may be primarily vascular, as in cerebral amyloid angiopathy, or in senile plaques and vascular.
One highly sensitive method is ELISA which is an immunosorbent assay which utilizes a pair of antibodies that recognize Amyloid beta.[83][84]
Atomic force microscopy, which can visualize nanoscale molecular surfaces, can be used to determine the aggregation state of Amyloid beta in vitro.[85]
Dual polarisation interferometry is an optical technique which can measure the very earliest stages of aggregration and inhibition by measuring the molecular size and densities as the fibrils elongate.[86][87] These aggregate processes can also be studied on lipid bilayer constructs.[88]
References
- 1 2 Vivekanandan S, Brender JR, Lee SY, Ramamoorthy A (Jul 2011). "A partially folded structure of amyloid-beta(1-40) in an aqueous environment". Biochemical and Biophysical Research Communications. 411 (2): 312–6. doi:10.1016/j.bbrc.2011.06.133. PMC 3148408
 . PMID 21726530.
. PMID 21726530. - ↑ Nussbaum JM, Seward ME, Bloom GS (Jan–Feb 2013). "Alzheimer disease: a tale of two prions". Prion. 7 (1): 14–9. doi:10.4161/pri.22118. PMC 3609044. PMID 22965142.
- ↑ Pulawski W, Ghoshdastider U, Andrisano V, Filipek S (Apr 2012). "Ubiquitous amyloids". Applied Biochemistry and Biotechnology. 166 (7): 1626–43. doi:10.1007/s12010-012-9549-3. PMC 3324686. PMID 22350870.
- ↑ Tharp WG, Sarkar IN (April 2013). "Origins of amyloid-β". BMC Genomics. 14 (1): 290. doi:10.1186/1471-2164-14-290. PMID 23627794.
- ↑ Hiltunen M, van Groen T, Jolkkonen J (2009). "Functional roles of amyloid-beta protein precursor and amyloid-beta peptides: evidence from experimental studies". Journal of Alzheimer's Disease. 18 (2): 401–12. doi:10.3233/JAD-2009-1154. PMID 19584429.
- ↑ Sadigh-Eteghad S, Talebi M, Farhoudi M, EJ Golzari S, Sabermarouf B, Mahmoudi J (2014). "Beta-amyloid exhibits antagonistic effects on alpha 7 nicotinic acetylcholine receptors in orchestrated manner". Journal of Medical Hypotheses and Ideas. 8: 48–52. doi:10.1016/j.jmhi.2014.01.001.
- ↑ Luo Y, Bolon B, Damore MA, Fitzpatrick D, Liu H, Zhang J, Yan Q, Vassar R, Citron M (Oct 2003). "BACE1 (beta-secretase) knockout mice do not acquire compensatory gene expression changes or develop neural lesions over time". Neurobiology of Disease. 14 (1): 81–8. doi:10.1016/S0969-9961(03)00104-9. PMID 13678669.
- ↑ Bogoyevitch MA, Boehm I, Oakley A, Ketterman AJ, Barr RK (Mar 2004). "Targeting the JNK MAPK cascade for inhibition: basic science and therapeutic potential". Biochimica et Biophysica Acta. 1697 (1-2): 89–101. doi:10.1016/j.bbapap.2003.11.016. PMID 15023353.
- ↑ Tabaton M, Zhu X, Perry G, Smith MA, Giliberto L (Jan 2010). "Signaling effect of amyloid-beta(42) on the processing of AβPP". Experimental Neurology. 221 (1): 18–25. doi:10.1016/j.expneurol.2009.09.002. PMC 2812589. PMID 19747481.
- ↑ Zou K, Gong JS, Yanagisawa K, Michikawa M (Jun 2002). "A novel function of monomeric amyloid beta-protein serving as an antioxidant molecule against metal-induced oxidative damage". The Journal of Neuroscience. 22 (12): 4833–41. PMID 12077180.
- ↑ Baruch-Suchodolsky R, Fischer B (May 2009). "Aβ40, either soluble or aggregated, is a remarkably potent antioxidant in cell-free oxidative systems". Biochemistry. 48 (20): 4354–70. doi:10.1021/bi802361k. PMID 19320465.
- ↑ Yao ZX, Papadopoulos V (Oct 2002). "Function of beta-amyloid in cholesterol transport: a lead to neurotoxicity". FASEB Journal. 16 (12): 1677–9. doi:10.1096/fj.02-0285fje. PMID 12206998.
- ↑ Igbavboa U, Sun GY, Weisman GA, He Y, Wood WG (Aug 2009). "Amyloid beta-protein stimulates trafficking of cholesterol and caveolin-1 from the plasma membrane to the Golgi complex in mouse primary astrocytes". Neuroscience. 162 (2): 328–38. doi:10.1016/j.neuroscience.2009.04.049. PMC 3083247. PMID 19401218.
- ↑ Maloney B, Lahiri DK (Nov 2011). "The Alzheimer's amyloid β-peptide (Aβ) binds a specific DNA Aβ-interacting domain (AβID) in the APP, BACE1, and APOE promoters in a sequence-specific manner: characterizing a new regulatory motif". Gene. 488 (1-2): 1–12. doi:10.1016/j.gene.2011.06.004. PMC 3381326. PMID 21699964.
- ↑ Bailey JA, Maloney B, Ge YW, Lahiri DK (Nov 2011). "Functional activity of the novel Alzheimer's amyloid β-peptide interacting domain (AβID) in the APP and BACE1 promoter sequences and implications in activating apoptotic genes and in amyloidogenesis". Gene. 488 (1-2): 13–22. doi:10.1016/j.gene.2011.06.017. PMC 3372404. PMID 21708232.
- ↑ Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, Burton MA, Goldstein LE, Duong S, Tanzi RE, Moir RD (2010). Bush AI, ed. "The Alzheimer's disease-associated amyloid beta-protein is an antimicrobial peptide". PLOS ONE. 5 (3): e9505. Bibcode:2010PLoSO...5.9505S. doi:10.1371/journal.pone.0009505. PMC 2831066. PMID 20209079.
- ↑ Kumar DK, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, Lefkowitz A, McColl G, Goldstein LE, Tanzi RE, Moir RD. "Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer's disease.". doi:10.1126/scitranslmed.aaf1059. PMID 27225182.
- ↑ "Alzheimer's culprit may fight other diseases". Science News.
- ↑ Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, Nedergaard M (Aug 2012). "A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β". Science Translational Medicine. 4 (147): 147ra111. doi:10.1126/scitranslmed.3003748. PMC 3551275. PMID 22896675.
- ↑ Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, O'Donnell J, Christensen DJ, Nicholson C, Iliff JJ, Takano T, Deane R, Nedergaard M (Oct 2013). "Sleep drives metabolite clearance from the adult brain". Science. 342 (6156): 373–7. Bibcode:2013Sci...342..373X. doi:10.1126/science.1241224. PMID 24136970.
- ↑ Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, Fieremans E, Axel L, Rusinek H, Nicholson C, Zlokovic BV, Frangione B, Blennow K, Ménard J, Zetterberg H, Wisniewski T, de Leon MJ (Aug 2015). "Clearance systems in the brain-implications for Alzheimer disease". Nature Reviews. Neurology. 11 (8): 457–70. doi:10.1038/nrneurol.2015.119. PMID 26195256.
- ↑ Sadigh-Eteghad S, Sabermarouf B, Majdi A, Talebi M, Farhoudi M, Mahmoudi J (2014). "Amyloid-beta: a crucial factor in Alzheimer's disease". Medical Principles and Practice. 24 (1): 1–10. doi:10.1159/000369101. PMID 25471398.
- ↑ Parker MH, Reitz AB (2000). "Assembly of β-Amyloid Aggregates at the Molecular Level". Chemtracts-Organic Chemistry. 13 (1): 51–56.
- ↑ Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ (Aug 2008). "Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory". Nature Medicine. 14 (8): 837–42. doi:10.1038/nm1782. PMC 2772133. PMID 18568035. Lay summary – Fox News.
- ↑ Prelli F, Castaño E, Glenner GG, Frangione B (Aug 1988). "Differences between vascular and plaque core amyloid in Alzheimer's disease". Journal of Neurochemistry. 51 (2): 648–51. doi:10.1111/j.1471-4159.1988.tb01087.x. PMID 3292706.
- 1 2 Zhao LN, Long H, Mu Y, Chew LY (2012). "The toxicity of amyloid β oligomers". International Journal of Molecular Sciences. 13 (6): 7303–27. doi:10.3390/ijms13067303. PMC 3397527. PMID 22837695.
- ↑ Arispe, N; Rojas, E; Pollard, H B (1993-01-15). "Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum.". Proceedings of the National Academy of Sciences of the United States of America. 90 (2): 567–571. doi:10.1073/pnas.90.2.567. ISSN 0027-8424. PMC 45704. PMID 8380642.
- ↑ Abramov, Andrey Y.; Canevari, Laura; Duchen, Michael R. (2004-12-06). "Calcium signals induced by amyloid β peptide and their consequences in neurons and astrocytes in culture". Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 8th European Symposium on Calcium. 1742 (1–3): 81–87. doi:10.1016/j.bbamcr.2004.09.006.
- ↑ Ekinci, Fatma J; Linsley, Maria-Dawn; Shea, Thomas B (2000-03-29). "β-Amyloid-induced calcium influx induces apoptosis in culture by oxidative stress rather than tau phosphorylation". Molecular Brain Research. 76 (2): 389–395. doi:10.1016/S0169-328X(00)00025-5.
- 1 2 3 Ghiso J, Frangione B (Dec 2002). "Amyloidosis and Alzheimer's disease". Advanced Drug Delivery Reviews. 54 (12): 1539–51. doi:10.1016/S0169-409X(02)00149-7. PMID 12453671.
- 1 2 Selkoe DJ (Oct 2001). "Clearing the brain's amyloid cobwebs". Neuron. 32 (2): 177–80. doi:10.1016/S0896-6273(01)00475-5. PMID 11683988.
- ↑ Hardy J, Duff K, Hardy KG, Perez-Tur J, Hutton M (Sep 1998). "Genetic dissection of Alzheimer's disease and related dementias: amyloid and its relationship to tau". Nature Neuroscience. 1 (5): 355–8. doi:10.1038/1565. PMID 10196523.
- ↑ Roses AD (Feb 1998). "Alzheimer diseases: a model of gene mutations and susceptibility polymorphisms for complex psychiatric diseases". American Journal of Medical Genetics. 81 (1): 49–57. doi:10.1002/(SICI)1096-8628(19980207)81:1<49::AID-AJMG10>3.0.CO;2-W. PMID 9514588.
- ↑ Xie L, Helmerhorst E, Taddei K, Plewright B, Van Bronswijk W, Martins R (May 2002). "Alzheimer's beta-amyloid peptides compete for insulin binding to the insulin receptor" (PDF). The Journal of Neuroscience. 22 (10): RC221. PMID 12006603.
- ↑ Ray WJ, Yao M, Mumm J, Schroeter EH, Saftig P, Wolfe M, Selkoe DJ, Kopan R, Goate AM (Dec 1999). "Cell surface presenilin-1 participates in the gamma-secretase-like proteolysis of Notch". The Journal of Biological Chemistry. 274 (51): 36801–7. doi:10.1074/jbc.274.51.36801. PMID 10593990.
- ↑ Roberts SB (Dec 2002). "Gamma-secretase inhibitors and Alzheimer's disease". Advanced Drug Delivery Reviews. 54 (12): 1579–88. doi:10.1016/S0169-409X(02)00155-2. PMID 12453675.
- ↑ Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M (Oct 1999). "Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE". Science. 286 (5440): 735–41. doi:10.1126/science.286.5440.735. PMID 10531052.
- ↑ Vassar R (Dec 2002). "Beta-secretase (BACE) as a drug target for Alzheimer's disease". Advanced Drug Delivery Reviews. 54 (12): 1589–602. doi:10.1016/S0169-409X(02)00157-6. PMID 12453676.
- ↑ Zlokovic BV, Frangione B (2003). Transport-clearance hypothesis for Alzheimer’s disease and potential therapeutic implications. Landes Bioscience. pp. 114–122.
- ↑ Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K (Jun 1985). "Amyloid plaque core protein in Alzheimer disease and Down syndrome". Proceedings of the National Academy of Sciences of the United States of America. 82 (12): 4245–9. Bibcode:1985PNAS...82.4245M. doi:10.1073/pnas.82.12.4245. PMC 397973. PMID 3159021.
- ↑ Castaño EM, Prelli F, Soto C, Beavis R, Matsubara E, Shoji M, Frangione B (Dec 1996). "The length of amyloid-beta in hereditary cerebral hemorrhage with amyloidosis, Dutch type. Implications for the role of amyloid-beta 1-42 in Alzheimer's disease". The Journal of Biological Chemistry. 271 (50): 32185–91. doi:10.1074/jbc.271.50.32185. PMID 8943274.
- ↑ Roher AE, Lowenson JD, Clarke S, Woods AS, Cotter RJ, Gowing E, Ball MJ (Nov 1993). "beta-Amyloid-(1-42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease". Proceedings of the National Academy of Sciences of the United States of America. 90 (22): 10836–40. Bibcode:1993PNAS...9010836R. doi:10.1073/pnas.90.22.10836. PMC 47873. PMID 8248178.
- ↑ Shinkai Y, Yoshimura M, Ito Y, Odaka A, Suzuki N, Yanagisawa K, Ihara Y (Sep 1995). "Amyloid beta-proteins 1-40 and 1-42(43) in the soluble fraction of extra- and intracranial blood vessels". Annals of Neurology. 38 (3): 421–8. doi:10.1002/ana.410380312. PMID 7668828.
- ↑ Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS (Dec 2001). "Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP". Nature. 414 (6864): 643–8. doi:10.1038/414643a. PMID 11740561.
- ↑ Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J (Sep 1999). "Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease". The American Journal of Pathology. 155 (3): 853–62. doi:10.1016/S0002-9440(10)65184-X. PMC 1866907. PMID 10487842.
- ↑ Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG (Apr 2003). "Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis". Science. 300 (5618): 486–9. Bibcode:2003Sci...300..486K. doi:10.1126/science.1079469. PMID 12702875.
- ↑ Gandy S, Simon AJ, Steele JW, Lublin AL, Lah JJ, Walker LC, Levey AI, Krafft GA, Levy E, Checler F, Glabe C, Bilker WB, Abel T, Schmeidler J, Ehrlich ME (Aug 2010). "Days to criterion as an indicator of toxicity associated with human Alzheimer amyloid-beta oligomers". Annals of Neurology. 68 (2): 220–30. doi:10.1002/ana.22052. PMC 3094694. PMID 20641005. Lay summary – Drug Discovery and Development.
- ↑ Laske, C., Leyhe, T., Stransky, E., Hoffmann, N., Fallgatter, A. J., & Dietzsch, J (2011). "Identification of a blood-based biomarker panel for classification of alzheimer's disease". The International Journal of Neuropsychopharmacology. 14: 1147–1155. doi:10.1017/S1461145711000459.
- ↑ Olsson F, Schmidt S, Althoff V, Munter LM, Jin S, Rosqvist S, Lendahl U, Multhaup G, Lundkvist J (Jan 2014). "Characterization of intermediate steps in amyloid beta (Aβ) production under near-native conditions". The Journal of Biological Chemistry. 289 (3): 1540–50. doi:10.1074/jbc.M113.498246. PMID 24225948.
- ↑ Hartmann T, Bieger SC, Brühl B, Tienari PJ, Ida N, Allsop D, Roberts GW, Masters CL, Dotti CG, Unsicker K, Beyreuther K (Sep 1997). "Distinct sites of intracellular production for Alzheimer's disease A beta40/42 amyloid peptides". Nature Medicine. 3 (9): 1016–20. doi:10.1038/nm0997-1016. PMID 9288729.
- ↑ Yin YI, Bassit B, Zhu L, Yang X, Wang C, Li YM (Aug 2007). "{gamma}-Secretase Substrate Concentration Modulates the Aβ42/Aβ40 Ratio: IMPLICATIONS FOR ALZHEIMER DISEASE". The Journal of Biological Chemistry. 282 (32): 23639–44. doi:10.1074/jbc.M704601200. PMID 17556361.
- ↑ Nalivaeva NN, Belyaev ND, Zhuravin IA, Turner AJ (2012). "The Alzheimer's amyloid-degrading peptidase, neprilysin: can we control it?". International Journal of Alzheimer's Disease. 2012: 383796. doi:10.1155/2012/383796. PMC 3412116. PMID 22900228.
- ↑ Maslow K (Mar 2008). "2008 Alzheimer's disease facts and figures". Alzheimer's & Dementia. 4 (2): 110–33. doi:10.1016/j.jalz.2008.02.005. PMID 18631956.
- ↑ Zhang S, Iwata K, Lachenmann MJ, Peng JW, Li S, Stimson ER, Lu Y, Felix AM, Maggio JE, Lee JP (Jun 2000). "The Alzheimer's peptide a beta adopts a collapsed coil structure in water". Journal of Structural Biology. 130 (2-3): 130–41. doi:10.1006/jsbi.2000.4288. PMID 10940221.
- ↑ Yang M, Teplow DB (Dec 2008). "Amyloid beta-protein monomer folding: free-energy surfaces reveal alloform-specific differences". Journal of Molecular Biology. 384 (2): 450–64. doi:10.1016/j.jmb.2008.09.039. PMC 2673916. PMID 18835397.
- ↑ Sgourakis NG, Merced-Serrano M, Boutsidis C, Drineas P, Du Z, Wang C, Garcia AE (Jan 2011). "Atomic-level characterization of the ensemble of the Aβ(1-42) monomer in water using unbiased molecular dynamics simulations and spectral algorithms". Journal of Molecular Biology. 405 (2): 570–83. doi:10.1016/j.jmb.2010.10.015. PMC 3060569. PMID 21056574.
- ↑ Sgourakis NG, Yan Y, McCallum SA, Wang C, Garcia AE (May 2007). "The Alzheimer's peptides Aβ40 and 42 adopt distinct conformations in water: a combined MD / NMR study". Journal of Molecular Biology. 368 (5): 1448–57. doi:10.1016/j.jmb.2007.02.093. PMC 1978067. PMID 17397862.
- ↑ Ahmed M, Davis J, Aucoin D, Sato T, Ahuja S, Aimoto S, Elliott JI, Van Nostrand WE, Smith SO (May 2010). "Structural conversion of neurotoxic amyloid-beta(1-42) oligomers to fibrils". Nature Structural & Molecular Biology. 17 (5): 561–7. doi:10.1038/nsmb.1799. PMC 2922021. PMID 20383142.
- ↑ Yu L, Edalji R, Harlan JE, Holzman TF, Lopez AP, Labkovsky B, Hillen H, Barghorn S, Ebert U, Richardson PL, Miesbauer L, Solomon L, Bartley D, Walter K, Johnson RW, Hajduk PJ, Olejniczak ET (Mar 2009). "Structural characterization of a soluble amyloid beta-peptide oligomer". Biochemistry. 48 (9): 1870–7. doi:10.1021/bi802046n. PMID 19216516.
- ↑ Strodel B, Lee JW, Whittleston CS, Wales DJ (Sep 2010). "Transmembrane structures for Alzheimer's Aβ(1-42) oligomers". Journal of the American Chemical Society. 132 (38): 13300–12. doi:10.1021/ja103725c. PMID 20822103.
- ↑ Mattson MP (Aug 2004). "Pathways towards and away from Alzheimer's disease". Nature. 430 (7000): 631–9. Bibcode:2004Natur.430..631M. doi:10.1038/nature02621. PMC 3091392. PMID 15295589.
- ↑ Citron M (Sep 2004). "Strategies for disease modification in Alzheimer's disease". Nature Reviews. Neuroscience. 5 (9): 677–85. doi:10.1038/nrn1495. PMID 15322526.
- ↑ Winblad B, Andreasen N, Minthon L, Floesser A, Imbert G, Dumortier T, Maguire RP, Blennow K, Lundmark J, Staufenbiel M, Orgogozo JM, Graf A (Jul 2012). "Safety, tolerability, and antibody response of active Aβ immunotherapy with CAD106 in patients with Alzheimer's disease: randomised, double-blind, placebo-controlled, first-in-human study". The Lancet. Neurology. 11 (7): 597–604. doi:10.1016/S1474-4422(12)70140-0. PMID 22677258. Lay summary – Karolinska Institutet.
- ↑ Wang CM, Devries S, Camboni M, Glass M, Martin PT (Sep 2010). "Immunization with the SDPM1 peptide lowers amyloid plaque burden and improves cognitive function in the APPswePSEN1(A246E) transgenic mouse model of Alzheimer's disease". Neurobiology of Disease. 39 (3): 409–22. doi:10.1016/j.nbd.2010.05.013. PMC 2913404. PMID 20493257.
- ↑ Baruch, Kuti; Deczkowska, Aleksandra; Rosenzweig, Neta; Tsitsou-Kampeli, Afroditi; Sharif, Alaa Mohammad; Matcovitch-Natan, Orit; Kertser, Alexander; David, Eyal; Amit, Ido (2016-02-01). "PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer's disease". Nature Medicine. 22 (2): 135–137. doi:10.1038/nm.4022. ISSN 1078-8956.
- ↑ Lashuel HA, Hartley DM, Balakhaneh D, Aggarwal A, Teichberg S, Callaway DJ (Nov 2002). "New class of inhibitors of amyloid-beta fibril formation. Implications for the mechanism of pathogenesis in Alzheimer's disease". The Journal of Biological Chemistry. 277 (45): 42881–90. doi:10.1074/jbc.M206593200. PMID 12167652.
- 1 2 Parker MH, Chen R, Conway KA, Lee DH, Luo C, Boyd RE, Nortey SO, Ross TM, Scott MK, Reitz AB (Nov 2002). "Synthesis of (-)-5,8-dihydroxy-3R-methyl-2R-(dipropylamino)-1,2,3,4-tetrahydronaphthalene: an inhibitor of beta-amyloid(1-42) aggregation". Bioorganic & Medicinal Chemistry. 10 (11): 3565–9. doi:10.1016/S0968-0896(02)00251-1. PMID 12213471.
- ↑ Finder VH, Vodopivec I, Nitsch RM, Glockshuber R (Feb 2010). "The recombinant amyloid-beta peptide Aβ1-42 aggregates faster and is more neurotoxic than synthetic Aβ1-42". Journal of Molecular Biology. 396 (1): 9–18. doi:10.1016/j.jmb.2009.12.016. PMID 20026079.
- ↑ "State of aggregation". Nature Neuroscience. 14 (4): 399. Apr 2011. doi:10.1038/nn0411-399. PMID 21445061.
- ↑ "BioPure Amyloid Peptides".
- ↑ Pappolla M, Bozner P, Soto C, Shao H, Robakis NK, Zagorski M, Frangione B, Ghiso J (Mar 1998). "Inhibition of Alzheimer beta-fibrillogenesis by melatonin". The Journal of Biological Chemistry. 273 (13): 7185–8. doi:10.1074/jbc.273.13.7185. PMID 9516407.
- ↑ Lahiri DK, Chen DM, Lahiri P, Bondy S, Greig NH (Nov 2005). "Amyloid, cholinesterase, melatonin, and metals and their roles in aging and neurodegenerative diseases". Annals of the New York Academy of Sciences. 1056: 430–49. Bibcode:2005NYASA1056..430L. doi:10.1196/annals.1352.008. PMID 16387707.
- ↑ Wang XC, Zhang YC, Chatterjie N, Grundke-Iqbal I, Iqbal K, Wang JZ (Jun 2008). "Effect of melatonin and melatonylvalpromide on beta-amyloid and neurofilaments in N2a cells". Neurochemical Research. 33 (6): 1138–44. doi:10.1007/s11064-007-9563-y. PMID 18231852.
- 1 2 3 4 5 Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, Fujiki N, Nishino S, Holtzman DM (Nov 2009). "Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle". Science. 326 (5955): 1005–7. Bibcode:2009Sci...326.1005K. doi:10.1126/science.1180962. PMC 2789838. PMID 19779148. Lay summary – News-Medical.Net.
- ↑ Pappolla MA, Bryant-Thomas TK, Herbert D, Pacheco J, Fabra Garcia M, Manjon M, Girones X, Henry TL, Matsubara E, Zambon D, Wolozin B, Sano M, Cruz-Sanchez FF, Thal LJ, Petanceska SS, Refolo LM (Jul 2003). "Mild hypercholesterolemia is an early risk factor for the development of Alzheimer amyloid pathology". Neurology. 61 (2): 199–205. doi:10.1212/01.wnl.0000070182.02537.84. PMID 12874399.
- ↑ Ramírez BG, Blázquez C, Gómez del Pulgar T, Guzmán M, de Ceballos ML (Feb 2005). "Prevention of Alzheimer's disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation". The Journal of Neuroscience. 25 (8): 1904–13. doi:10.1523/JNEUROSCI.4540-04.2005. PMID 15728830.
- ↑ Milton NG (Oct 2002). "Anandamide and noladin ether prevent neurotoxicity of the human amyloid-beta peptide". Neuroscience Letters. 332 (2): 127–30. doi:10.1016/S0304-3940(02)00936-9. PMID 12384227.
- ↑ Refolo LM, Malester B, LaFrancois J, Bryant-Thomas T, Wang R, Tint GS, Sambamurti K, Duff K, Pappolla MA (Aug 2000). "Hypercholesterolemia accelerates the Alzheimer's amyloid pathology in a transgenic mouse model". Neurobiology of Disease. 7 (4): 321–31. doi:10.1006/nbdi.2000.0304. PMID 10964604.
- ↑ Refolo LM, Pappolla MA, LaFrancois J, Malester B, Schmidt SD, Thomas-Bryant T, Tint GS, Wang R, Mercken M, Petanceska SS, Duff KE (Oct 2001). "A cholesterol-lowering drug reduces beta-amyloid pathology in a transgenic mouse model of Alzheimer's disease". Neurobiology of Disease. 8 (5): 890–9. doi:10.1006/nbdi.2001.0422. PMID 11592856.
- 1 2 Lee JY, Cole TB, Palmiter RD, Suh SW, Koh JY (May 2002). "Contribution by synaptic zinc to the gender-disparate plaque formation in human Swedish mutant APP transgenic mice". Proceedings of the National Academy of Sciences of the United States of America. 99 (11): 7705–10. Bibcode:2002PNAS...99.7705L. doi:10.1073/pnas.092034699. PMC 124328. PMID 12032347.
- ↑ Schneider JS, Pioli EY, Jianzhong Y, Li Q, Bezard E (Apr 2013). "Effects of memantine and galantamine on cognitive performance in aged rhesus macaques". Neurobiology of Aging. 34 (4): 1126–32. doi:10.1016/j.neurobiolaging.2012.10.020. PMID 23158762.
- ↑ Volicer L, Harper DG, Manning BC, Goldstein R, Satlin A (May 2001). "Sundowning and circadian rhythms in Alzheimer's disease". The American Journal of Psychiatry. 158 (5): 704–11. doi:10.1176/appi.ajp.158.5.704. PMID 11329390.
- ↑ Schmidt SD, Nixon RA, Mathews PM (2012). "Tissue processing prior to analysis of Alzheimer's disease associated proteins and metabolites, including Aβ". Methods in Molecular Biology. Methods in Molecular Biology. 849: 493–506. doi:10.1007/978-1-61779-551-0_33. ISBN 978-1-61779-550-3. PMID 22528111.
- ↑ Schmidt SD, Mazzella MJ, Nixon RA, Mathews PM (2012). "Aβ measurement by enzyme-linked immunosorbent assay". Methods in Molecular Biology. Methods in Molecular Biology. 849: 507–27. doi:10.1007/978-1-61779-551-0_34. ISBN 978-1-61779-550-3. PMID 22528112.
- ↑ Stine WB, Dahlgren KN, Krafft GA, LaDu MJ (Mar 2003). "In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis". The Journal of Biological Chemistry. 278 (13): 11612–22. doi:10.1074/jbc.M210207200. PMID 12499373.
- ↑ Gengler S, Gault VA, Harriott P, Hölscher C (Jun 2007). "Impairments of hippocampal synaptic plasticity induced by aggregated beta-amyloid (25-35) are dependent on stimulation-protocol and genetic background". Experimental Brain Research. 179 (4): 621–30. doi:10.1007/s00221-006-0819-6. PMID 17171334.
- ↑ Rekas A, Jankova L, Thorn DC, Cappai R, Carver JA (Dec 2007). "Monitoring the prevention of amyloid fibril formation by alpha-crystallin. Temperature dependence and the nature of the aggregating species". The FEBS Journal. 274 (24): 6290–304. doi:10.1111/j.1742-4658.2007.06144.x. PMID 18005258.
- ↑ Sanghera N, Swann MJ, Ronan G, Pinheiro TJ (Oct 2009). "Insight into early events in the aggregation of the prion protein on lipid membranes". Biochimica et Biophysica Acta. 1788 (10): 2245–51. doi:10.1016/j.bbamem.2009.08.005. PMID 19703409.
Further reading
- Martins IC, Kuperstein I, Wilkinson H, Maes E, Vanbrabant M, Jonckheere W, Van Gelder P, Hartmann D, D'Hooge R, De Strooper B, Schymkowitz J, Rousseau F (Jan 2008). "Lipids revert inert Aβ amyloid fibrils to neurotoxic protofibrils that affect learning in mice". The EMBO Journal. 27 (1): 224–33. doi:10.1038/sj.emboj.7601953. PMC 2206134. PMID 18059472.
- Istrate AN, Tsvetkov PO, Mantsyzov AB, Kulikova AA, Kozin SA, Makarov AA, Polshakov VI (Jan 2012). "NMR solution structure of rat aβ(1-16): toward understanding the mechanism of rats' resistance to Alzheimer's disease". Biophysical Journal. 102 (1): 136–43. Bibcode:2012BpJ...102..136I. doi:10.1016/j.bpj.2011.11.4006. PMC 3250693. PMID 22225807.